温馨提示:请使用火狐或者Chrome的网页浏览器来查看报告
微科盟细菌基因组完成图分析结题报告
一 概述
细菌基因组完成图是采用三代测序平台,一次性获得完整的细菌基因组图谱,实现没有gap和N碱基的组装,同时可以获得质粒基因组信息。二代测序技术难以解决细菌基因组中重复区域与高GC区域组装的问题,因此往往需要借助PCR扩增、Sanger测序等方式填补组装后基因组中的gap区域。而基于三代测序技术充分发挥了超长读长的优势,轻松跨越细菌基因组中的这些复杂结构,不但在组装流程上更为简洁,还能够对组装结果更加可靠稳定。
二 项目流程
2.1 测序实验流程
DNA提取及检测 采用SDS或STE方法对样本的基因组DNA进行提取,然后用琼脂糖凝胶电泳检测DNA的纯度和完整性,用Qubit进行定量。
经电泳检测合格的DNA样品用Covaris超声波破碎仪随机打断成长度约为350bp的片段。处理完成后的DNA片段,使用NEBNext®Ultra™ DNA Library Prep KitforIllumina(NEB, USA)试剂盒,经末端修复、加A尾、加测序接头、纯化、PCR扩增等步骤完成整个文库制备。文库构建完成后,先使用Qubit 2.0进行初步定量,稀释文库至2ng/ul,随后使用Agilent 2100对文库的插入片段进行检测,insert size符合预期后,使用q-PCR方法对文库的有效浓度进行准确定量,以保证文库质量。
构建测序文库后将经电泳检测合格的DNA样品进行纯化,并将构建好的文库经Qubit浓度定量,并利用Agilent 2100检测插入片段大小,最后用Nanopore平台进行测序。

图2-1 细菌基因组完成图样品制备流程图
图2-2 细菌基因组完成图样品测序类型
2.2 生信分析流程
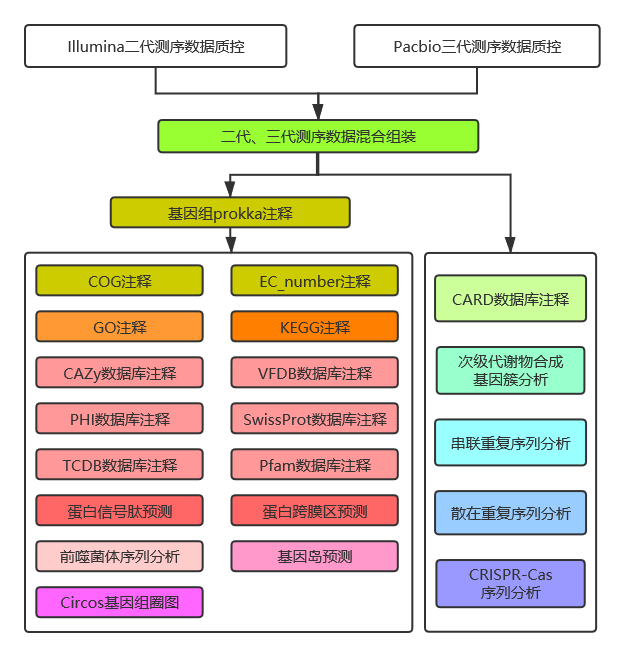
图2-3 生信分析流程图
详细过程见结果解读部分。
三 结果文件夹解读
3.1 质控
01.Data_qc/ ├──illumina_fastp.xls [二代测序数据质控结果] └──pacbio_sum.xls [三代测序数据longQC质控结果]
高通量测序得到的图像经 Base Calling 转化为原始测序序列(Reads),我们称之为 Raw Data 或 Raw Reads,结果以 FASTQ(简写为fq)文件格式储存,它包含测序序列(Reads)的序列信息及其对应的测序质量信息。PE 文库的数据结果中,每个样品均有测序两端的 Reads,并且 Reads 的顺序是严格一致、相互对应的。 FASTQ 文件中每条 read 都由四行构成,文件格式如下:

图1-1-1 FASTQ格式文件内容示例
其中,第 1 行和第 3 行为 read 名称(后来为了节省储存空间省略掉第 3 行“+”后面的序列名称),由 Illumina 测序仪产生;第 2 行是 read 的碱基序列,第 4 行是 read 中每个碱基对应的测序质量分数,由 Base Calling 转化而来,每个字母对应的 ASCII 值减去相应测序质量系统的 Phred quality score(33 或 64),即为该碱基的测序质量值(Phred 值)。
在获得每个样品二代测序数据之后,首先需要对测序的数据质量进行评估并对低质量的数据进行去除,以保证后续分析结果的可信度,以上步骤称之为原始数据的质量控制。质量控制获得的高质量序列则用于下游的数据分析。质控流程采用软件fastp[2],具体处理步骤如下:
- 去除带接头(adapter)的 paired reads;
- 当单端测序read 中含有的低质量(sQ <= 20)碱基数超过该条read碱基总数的20%时,去除此 paired reads;
- 去除PCR扩增产生的重复reads。
测序数据质控前后的质量信息统计表格如下:
表1-1-1 原始数据的质量信息统计表格(illumina_fastp.xls)
| sampleID | Raw total reads | Raw total bases(Mb) | Raw Q20 rate | Raw Q30 rate | Clean total reads | Clean total bases(Mb) | Clean Q20 rate | Clean Q30 rate | Clean gc content(%) | Filtered Reads(%) |
|---|---|---|---|---|---|---|---|---|---|---|
| C1 | 5956966 | 893.54M | 96.92 | 91.59 | 5954198 | 862.1M | 97.12 | 91.83 | 51.12 | 96.48 |
| C2 | 5956966 | 893.54M | 96.92 | 91.59 | 5954198 | 862.1M | 97.12 | 91.83 | 51.12 | 96.48 |
| Z1 | 10825608 | 1623.84M | 96.96 | 91.70 | 10820610 | 1564.93M | 97.21 | 92.00 | 40.46 | 96.37 |
- sampleID: 样本ID
- Raw total reads: 原始测序数据reads数
- Raw total bases(Mb): 样本碱基总数
- Raw Q20 rate: 质控前质量分测序高于20(错误率0.01)的碱基占比(Q20)
- Raw Q30 rate: 质控前质量分测序高于30(错误率0.001)的碱基占比(Q30)
- Clean total reads:质控后测序数据reads数
- Clean total bases(Mb):质控后测序数据碱基数
- Clean Q20 rate:质控后测序质量Q20比例
- Clean Q30 rate:质控后测序质量Q30比例
- Clean gc content(%):质控后测序数据GC含量
- Filtered Reads(%):质控后保留碱基数比例
三代测序平台Pacbio Sequel II测序仪的下机数据,我们称之为subreads,它们是已经去除了adapter和barcode序列的数据。
我们用bam2fastq[1]软件,从测序仪下机数据subreads.bam文件提取出fastq格式的数据文件,里面所有碱基的碱基质量值都为0,这是Pacbio Sequel II平台的测序特点,即由于出现测序错误的碱基几乎是随机分布的,不像Illumina测序那样具有位置倾向性,无法提供有实际意义的单个碱基的测序质量值。
我们这里的质控处理,主要是用fastp[2]软件把长度偏小的subreads给剔除掉,而不做局部的序列裁剪。在三代测序文库的构建中,一般每一条待测的生物样本序列会在10kb以上,如果subread测出来的序列太短,则有可能不是想测的生物样本序列,而是实验过程中引入的其它杂质序列。所以我们在质控步骤中把长度小于0.5kb的subreads给剔除掉,这样保留下来的数据我们称之为clean data。
最后我们用LongQC[3]软件对clean data做质控分析报告。
表1-1-2 三代序列质控后的结果统计表(pacbio_sum.xls)
| sampleID | Raw total reads | Raw total bases(Mb) | Clean total reads | Clean total bases(Mb) | Clean gc content(%) | Filtered Reads(%) | Longest read length(bp) | Estimated non-sense read fraction |
|---|---|---|---|---|---|---|---|---|
| C1 | 378760 | 4146.96M | 374977 | 4145.98M | 49.90 | 99.98 | 191675 | 0.0044 |
| C2 | 378760 | 4146.96M | 374977 | 4145.98M | 49.90 | 99.98 | 191675 | 0.0044 |
| Z1 | 487233 | 6496.48M | 483693 | 6495.47M | 39.71 | 99.98 | 178945 | 0.0030 |
- sampleID: 样本ID
- Raw total reads:原始测序数据reads数
- Raw total bases(Mb):原始测序数据碱基数
- Clean total reads:质控后测序数据reads数
- Clean total bases(Mb):质控后测序数据碱基数
- Clean gc content(%):质控后测序数据GC含量
- Filtered Reads(%):质控后保留碱基数比例
- Longest read length(bp):最长reads的长度
- Estimated non-sense read fraction:评估出来的non-sense reads所占比例,应该少于0.3。
比例高表明: 1. 测序数据有问题 2. 覆盖度不高
3.2 基因组组装
02.Assemble/ ├── all_assembly_stats.xls[最终组装结果统计结果] ├── all_sample_gc_depth_stat.xls[基因组gc_depth估计结果统计表] ├── all_sample_genomesize_stat.xls[基因组大小估计结果统计表] ├── all_samples_stats.xls [最终组装结果统计表] ├──* │ ├──*.all.plassmid.blast.summary.top5[质粒注释结果,若无质粒,则无此文件] │ ├──*.final.assembly.Chromosome.fasta[染色体序列] │ ├──*.final.assembly.fasta[最终组装序列,包含质粒和染色体] │ ├──*.final.Plasmid.fasta[最终质粒序列,若无质粒,则无此文件] │ ├──*.final.stat.xls[统计文件] │ ├── genome_gc_depth[GC-Depth分析文件夹] │ │ ├──*.gc_depth_describe.txt[GC-Depth分析及污染评估的文字描述] │ │ ├──*.gc_depth.pdf[GC-Depth图片,pdf格式] │ │ └──*.gc_depth.png[GC-Depth图片,png格式] │ └── genome_size[基因组大小评估分析文件夹] │ ├── plot.png[基因组大小评估分析文件结果图] │ └── summary.txt[基因组大小评估分析文件夹结果]
使用unicycler[5]软件(默认参数:--keep 0 --min_fasta_length 1000),以质控后的clean data作为输入数据,做基因组组装。
组装出来的基因组序列文件请查看*final.assembly.fasta文件。
为了鉴别组装出来是质粒还是染色体,使用RFPlasmid[6]软件(默认参数:--species Generic --jelly)来区分质粒和染色体。对于质粒序列,将组装的质粒序列与质粒数据库进行比对,对比对结果进行分析,total score值排前5的比对结果见相关文件。对于每个样本最终得到的contigs, 我们使用quast[4]软件进行质量评估。
详细结果说明见组装结果说明。
表2-1-1 样品基因组组装结果统计(all_samples_stats.xls )
| sampleID | contigs | Total contig length | Contig N50 | Largest contig | GC content % |
|---|---|---|---|---|---|
| C1 | 3 | 4934836 | 4782682 | 4782682 | 50.69 |
| C2 | 3 | 4934836 | 4782682 | 4782682 | 50.69 |
| Z1 | 1 | 4041531 | 4041531 | 4041531 | 39.12 |
- Sample ID: 样本ID
- Type:类型,质粒或染色体;
- Contig ID:序列ID;
- Size(bp):序列长度
- GC%:GC含量
- Circular:是否环化
注:有些细菌组装出来多条染色体,可能原因有
- 1.该菌株确实有多条染色体(或巨质粒);
- 2.该菌株是复杂基因组;
- 基因组大小 >7M;染色体基因组个数 + 质粒个数 > 4;
转座子或其它重复序列在基因组中的比例异常高 ( > 10% );GC含量异常高等可以称为复杂基因组 - 3.该菌株有污染等
基因组大小是指单倍体细胞核中的所含的DNA的总量。预测未知基因组大小的方法可以通过 Illumina 测序数据的 k-mer 分析进行估计。
我们使用利用Jellyfish[8]对reads进行处理,获得不同频率的k-mer信息,然后利用相应的软件进行基因组大小预估。而Kmerfreq利用自身算法进行K-mer剪切(K-mer=17)统计,利用K-mer频率分布使用GenomeScope[9]进行基因组大小预估。
表2-2-1 基因组大小估计结果统计表(all_sample_genomesize_stat.xls)
| sampleID | Heterozygosity | Genome_Haploid_Length |
|---|---|---|
| C1 | 0.08 | 4970504.0 |
| C2 | 0.08 | 4970504.0 |
| Z1 | 0.37 | 3635336.0 |
- sampleID: 样本ID
- Heterozygosity:杂合率
- Genome_Haploid_Length(bp):基因组大小
对组装的基因组序列以500bp为滑窗窗口(windows),计算滑窗窗口的平均GC含量和平均深度并作图。基于每一个windows对应的平均GC和平均深度进行绘图。可以根据此图分析测序数据是否存在GC偏向性以及样本是否存在污染。
我们使用bwa[7]和相应脚本对组装后的基因组序列和质控后测序序列进行分析,详细统计结果见下表:
表2-2-2 基因组GC_depth分析结果(all_sample_gc_depth_stat.xls)
| sampleID | ill_depth | ill_reslut |
|---|---|---|
| C1 | 175 | 样本无明显污染 |
| C2 | 175 | 样本无明显污染 |
| Z1 | 438 | 样本无明显污染 |
- sampleID: 样本ID
- ill_depth:二代测序数据测序深度
- ill_reslut:二代测序数据GC_depth分析结果
3.3 基因预测
03.Gene_predict/
├──all_sample.prokka.stat.txt [所有样本prokka注释结果]
├──all_ncRNA.stat.xls [ncRNA结果统计表]
├── COG
│ ├── *.cog.txt [COG比对结果]
│ └── plot [COG图片]
│ ├── cog_summed_up_summary_cog_color.pdf[COG汇总图片]
│ ├── cog_summed_up_summary_cog_color.png[COG汇总图片]
│ ├── cog_summed_up_summary_ggplot2_color.pdf[COG汇总图片]
│ └── cog_summed_up_summary_ggplot2_color.png[COG汇总图片]
├── EC_number
│ └── *.ec_number.txt [ec_number结果]
├── ncRNA
│ ├── *.denovo.rRNA.fa [rRNA预测结果,fasta格式]
│ ├── *.ncRNA.stat.xls [ncRNA 预测结果统计表]
│ ├── *.rRNA.gff [rRNA预测结果,gff格式]
│ ├── *.srna.gff [sRNA预测结果,gff格式]
│ ├── *.tRNA.gff3[tRNA预测结果,gff格式]
│ └── *.tRNA.structure[tRNA结构预测结果]
└── prokka
├── *.faa[prokka预测的蛋白氨基酸序列]
├── *.ffn[prokka注释的核苷酸序列]
├── *.fna[用于提交的Contig序列(核苷酸)]
├── *.fsa【用于提交的Contig序列(核苷酸)]
├── *.gbk[genbank格式的注释文件]
├── *_gene_length.png[基因长度统计图]
├── *.gff[包含序列和注释的GFF文件]
├── *.pdf [注释汇总统计图]
├── *.png[注释汇总统计图]
├── *.sqn[用于提交的Sequin可编辑文件]
├── *_summarize.txt[注释汇总统计,用于绘图]
├── *.tbl[用于提交的特征表(Feature table)]
├── *.tsv[注释基因列表]
└── *.txt[注释汇总统计]
3.3.1 基因预测
使用细菌基因组功能注释工具prokka,对组装生成的assembly.fasta基因组序列文件,做基因预测和功能注释,然后用R语言分别对不同的功能注释做统计。
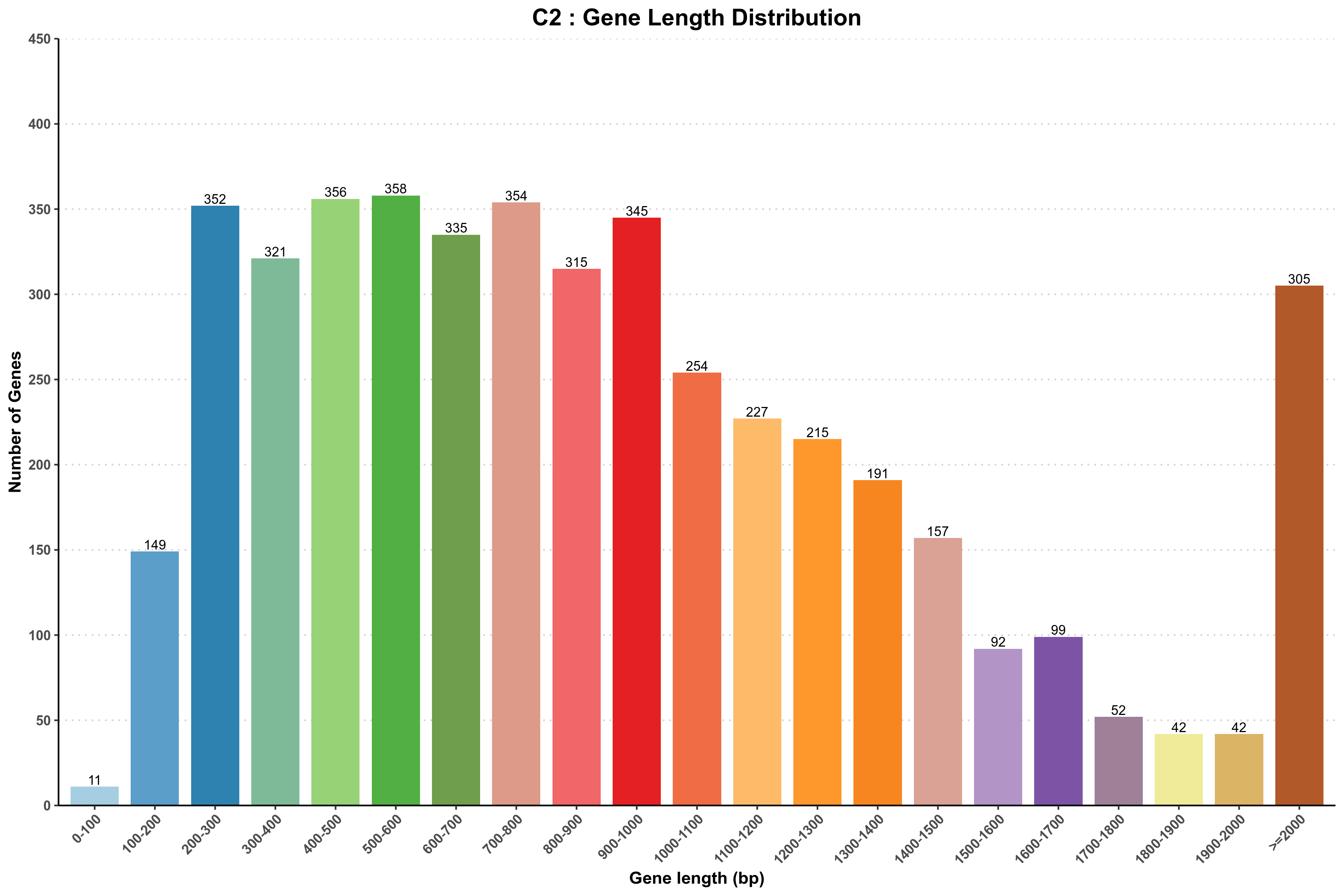
图3-1-1 样品基因长度分布图
说明:横坐标为基因长度,纵坐标为相应基因个数
表3-1-1 基因组prokka注释功能类型统计表(all_sample.prokka.stat.txt)
| ContigID | NumOf_ftype | NumOf_CDS | NumOf_repeat_region | NumOf_rRNA | NumOf_tmRNA | NumOf_tRNA | NumOf_gene | NumOf_COG | NumOf_EC_number |
|---|---|---|---|---|---|---|---|---|---|
| C1 | 4682 | 4572 | 1.0 | 22 | 1 | 86 | 3473 | 2840 | 1641 |
| C2 | 4682 | 4572 | 1.0 | 22 | 1 | 86 | 3473 | 2840 | 1641 |
| Z1 | 3760 | 3651 | NaN | 22 | 1 | 86 | 2471 | 2038 | 1221 |
- ContigID:样本名
- NumOf_ftype:所有功能类型数目
- NumOf_CDS:编码基因数目
- NumOf_repeat_region:重复区域数目
- NumOf_rRNA:rRNA数目
- NumOf_tmRNA:tmRNA数目
- NumOf_gene:表示注释到具体的基因英文单词数目
- NumOf_COG:cog数目
- NumOf_EC_number:EC数目
3.3.2 COG预测
COG,即Clusters of Orthologous Groups of proteins(直系同源蛋白簇)。COG是由NCBI创建并维护的蛋白数据库,根据细菌、藻类和真核生物完整基因组的编码蛋白系统进化关系分类构建而成。每一簇COG由直系同源蛋白序列构成,通过比对可以将某个蛋白序列归到某一个已知功能的COG中,从而可以推测该未知功能的蛋白的功能信息。 我们对基因组prokka注释结果中的.tsv文件,提取COG注释信息,然后用R语言做分类统计。
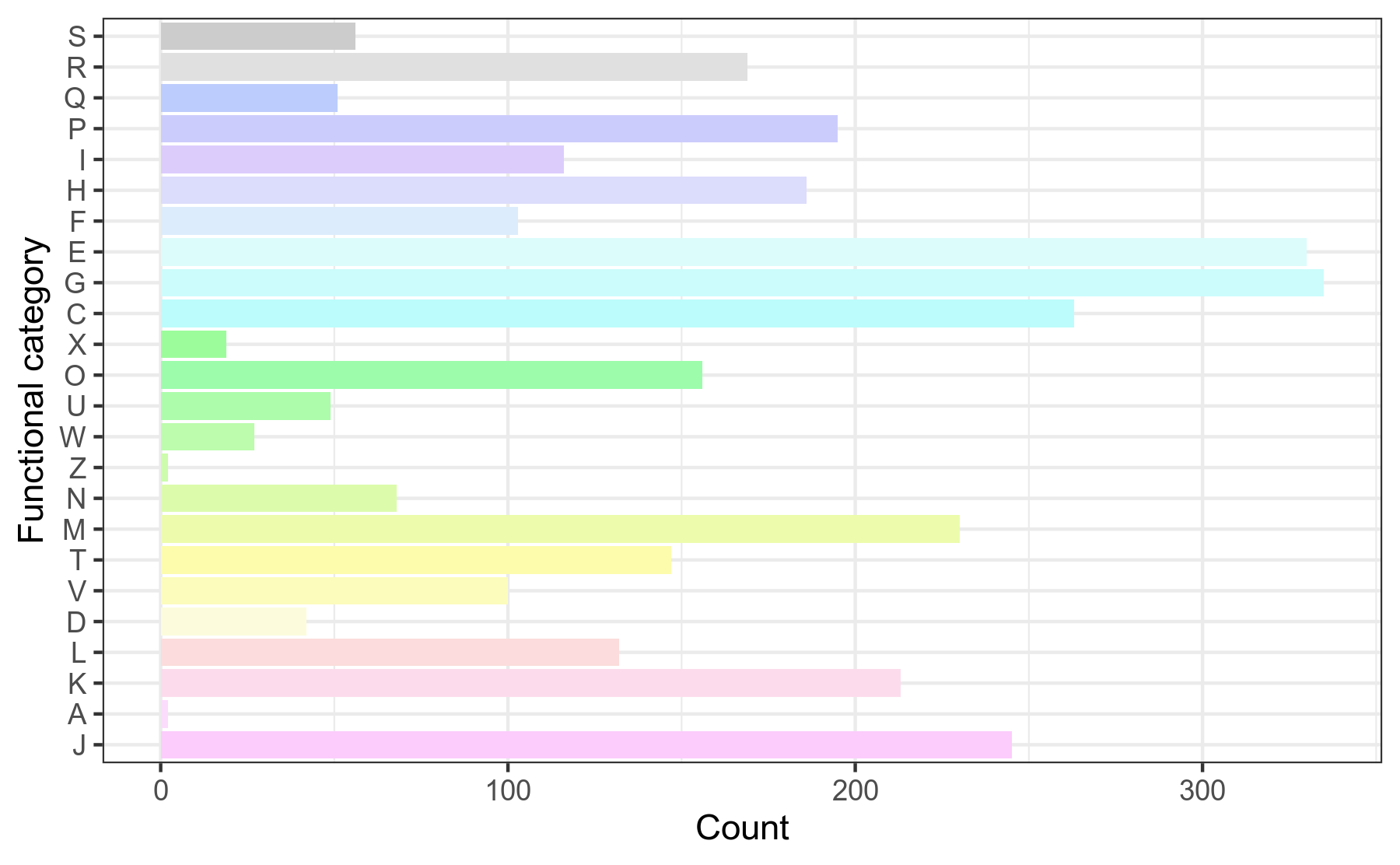
图3-1-2 COG注释功能分类统计图
说明:横坐标为相应基因个数,纵坐标为COG分类
COG分类字母代表含义见COG分类结果
3.3.3 EC_number预测
EC_number是酶学委员会(Enzyme Commission)为酶所设计的一套编号分类法,每一个酶的编号都以字母“EC”开头,接着以四段号码来表示,这些号码表示对酶作出不同层级的分类。例如三肽胺基-蛋白酶的编号为EC3.4.11.4,当中的“EC3”是指水解酶(即需要用水来将其它分子分解的酶);“EC3.4”是指与肽键作用的水解酶;“EC3.4.11”是指从多肽中分开胺基末端的水解酶;“EC3.4.11.4”则是指从三肽中分开胺基末端的水解酶。 我们对基因组prokka注释结果中的.tsv文件,提取EC_number注释信息,整理成单独的表格文件.
3.3.4 非编码RNA预测
非编码RNA(ncRNA)是一类执行多种生物学功能的RNA分子,其本身并不携带翻译为蛋白质的信息,直接在RNA水平对生命活动发挥作用。对于微生物而言,研究较为普遍的包括sRNA、rRNA、tRNA。
tRNA:转运RNA(Transfer RNA),又称传送核糖核酸、转移核糖核酸,通常简称为tRNA,是一种由76-90个核苷酸所组成的RNA,其3'端可以在氨酰-tRNA合成酶催化之下,接附特定种类的氨基酸。转译的过程中,tRNA可借由自身的反密码子识别mRNA上的密码子,将该密码子对应的氨基酸转运至核糖体合成中的多肽链上。本分析中通过tRNAscan-SE软件对tRNA进行预测。
rRNA:即核糖体RNA,rRNA在相邻物种中高度保守。rRNA的预测方法有两种,一是通过与近缘参考序列的rRNA库比对找到rRNA,二是用rRNAmmer软件预测rRNA。
sRNA:小RNA,首先进行Rfam database比对注释,接着用cmsearch程序(参数默认)确定最终的sRNA。
snRNA:(small nuclearRNA,小核RNA),它是真核生物转录后加工过程中RNA剪接体(spilceosome)的主要成分。
miRNA:MicroRNA(miRNA)是在真核生物中发现的一类内源性的具有调控功能的非编码RNA,前体全长约90bp,其成熟miRNA大小长约20~25个核苷酸(nt)。miRNA广泛存在于真核生物中,是一组不编码蛋白质的短序列RNA,它本身不具有开放阅读框(ORF),对基因的表达具有调控作用。
sRNA、snRNA、miRNA的预测原理类似,首先用Rfam软件进行Rfam database比对注释,接着用其cmsearch程序(参数默认)确定最终的sRNA、snRNA、miRNA。由于细菌是原核生物,故ncRNA 的类型主要指 tRNA、rRNA及 sRNA 三种。
此次分析中的tRNA和rRNA预测的结果可能和prokka基因预测的结果不同,这是由于采用的软件不同导致的。
详细结果说明见ncRNA注释结果
各样本ncRNA结果统计表格如下:
表3-1-2 各样本ncRNA结果统计表格统计表(all_ncRNA.stat.xls)
| sampleID | type | count | total_length | average_length |
|---|---|---|---|---|
| C1 | tRNA | 85 | 6330 | 74.47 |
| C1 | 16s_rRNA | 7 | 10703 | 1529.00 |
| C1 | 23s_rRNA | 7 | 20302 | 2900.29 |
| C1 | 5s_rRNA | 8 | 912 | 114.00 |
| C1 | sRNA | 118 | 12487 | 105.82 |
| C2 | tRNA | 85 | 6330 | 74.47 |
| C2 | 16s_rRNA | 7 | 10703 | 1529.00 |
| C2 | 23s_rRNA | 7 | 20302 | 2900.29 |
| C2 | 5s_rRNA | 8 | 912 | 114.00 |
| C2 | sRNA | 118 | 12487 | 105.82 |
| Z1 | tRNA | 84 | 6242 | 74.31 |
| Z1 | 16s_rRNA | 7 | 10710 | 1530.00 |
| Z1 | 23s_rRNA | 7 | 20775 | 2967.86 |
| Z1 | 5s_rRNA | 8 | 912 | 114.00 |
| Z1 | sRNA | 19 | 2255 | 118.68 |
- samplieID:样本名
- type:所有功能类型数目
- count:拷贝数目
- total_length:该种类非编码 RNA 的总长度,单位 bp
- average_length:该种类非编码 RNA 的平均长度,单位 bp(base pair,碱基对)
3.4 菌种鉴定
04.Bac_is_species/ ├── all_bac_taxid_16s.txt [所有样本16s统计结果] ├── 16s_ani_sum.tsv [所有样本基因组16s与ani结果] ├── * │ └── 16s_ANI [ANI结果文件夹] │ ├── aai_heatmap.svg [aai结果图] │ ├── aai_matrix.tsv [aai矩阵表] │ ├── aai_summary.tsv[aai汇总表] │ ├── ANIb_percentage_identity.pdf[ani汇总图] │ ├── ANIb_percentage_identity.png[ani汇总图] │ ├── ANIb_percentage_identity.tab[ani汇总表] │ ├── TETRA_correlations.pdf[TETRA汇总图] │ ├── TETRA_correlations.png[TETRA汇总图] │ └── TETRA_correlations.tab[TETRA汇总表]
3.4.1 16s序列物种鉴定
使用Prokka预测的所有16s序列结果与SILVA-16S数据库进行blastn得到结果。SILVA数据库有超过10万个手动检查的序列,是细菌分类经典数据库。16S rRNA进行blastn推荐物种界限是98.6%,这里我们使用的为99%。
另外,如果有16S序列起源于超过1物种,则被视为受污染的基因组。
详细结果说明见细菌鉴定结果说明。
表4-1-1 16s物种鉴定表格(all_bac_taxid_16s.txt)
| sampleID | 16S_id | 16S_start | 16S_end | species_name | taxid |
|---|---|---|---|---|---|
| C1 | CP013663 | 4772352 | 4773907 | Escherichia_coli | 562 |
| C2 | CP013663 | 4772352 | 4773907 | Escherichia_coli | 562 |
| Z1 | KF928778 | 1 | 1542 | Proteus_mirabilis | 584 |
- sampleID: 样本ID
- 16S_id:16s序列id
- 16S_start: 16S序列在染色体开始位置(bp)
- 16S_end: 16S序列在染色体结束位置(bp)
- species_name: 16s序列鉴定得出的物种名
- taxid: NCBI物种分类号
3.4.2 ANI序列物种鉴定
ANI(average nucleotide identity)、AAI(amino acid identity)和TETRA(tetra-nucleotide signature)是计算比较基因组学常用算法,可被用于区分物种。ANI是平均核苷酸相似度,是在核苷酸水平比较两个基因组亲缘关系的指标,其在近缘物种之间有较高的区分度。类似的,AAI是氨基酸一致性,是在氨基酸水平比较两个基因组亲缘关系的指标。而TETRA是统计四核苷酸序列(tetra-nucleotide)的频率。当两个基因组序列相似时,这些四核苷酸频率的相关性越高。因此,两个基因组序列之间四核苷酸频率相关性可以粗略的用于确定两个基因组的基因组相关性。 三者物种界限推荐为:95%(ANI)、95%(AAI)和99%(TERAE).
为了进一步鉴定细菌物种,使用物种分类号(默认:16s物种分类)从NCBI上下载同分类号的基因组序列(下载最多十个)。将组装好的序列与下载的基因组进行比较ANI,AAI和TETRA,以确定组装出来的基因组物种归属。 ANI和TETRA是通过pyani[12]得到的(使用默认参数:-m ANIb 和-m TETRA),AAI是通过CompareM[11]得到的(使用默认参数:aai_wf)
表4-2-1 16s物种鉴定表格(16s_ani_sum.tsv)
| sampleID | 16s_median_ANI(%) | 16s_median_AAI(%) | 16s_median_TETRA(%) | 16s_max_ANI_strians(max_ANI_score(%)) | 16s_is_same_species | species_name | taxid |
|---|---|---|---|---|---|---|---|
| C1 | 98.48 | 98.68 | 99.92 | ASM30897v2.E.coli.genomic(98.92) | True | Escherichia_coli | 562 |
| C2 | 98.48 | 98.68 | 99.92 | ASM30897v2.E.coli.genomic(98.92) | True | Escherichia_coli | 562 |
| Z1 | 99.14 | 99.20 | 99.93 | ASM78387v2.P.mira.FDAARGOS(99.35) | True | Proteus_mirabilis | 584 |
- sampleID: 样本ID
- 16s_median_ANI(%): ANI 结果中位数
- 16s_median_AAI(%):AAI 结果中位数
- 16s_median_TETRA(%):TETRA结果中位数
- 16s_max_ANI_strians(max_ANI_score(%)):ANI结果最大的菌株名(ANI得分)
- 16s_is_same_species:与16s结果是否一致
- species_name: 16s序列鉴定得出的物种名
- taxid: NCBI物种分类号
- 注:菌株名其名称构成为Reference Sequence ID+ 物种名+菌株名;与16s结果是否一致按照max_ANI_score(%)是否大于95判断,若大于95则是True。此结果只做一个参考,不做最终结果.
3.5 基因组组分分析
3.5.1 噬菌体分析
Prophage/
├── all_sample.prophage.xls[所有样本噬菌体结果]
└── *
├── *_phage.fasta【噬菌体结果文件,fasta格式】
├── *_prophage_coordinates.tsv【噬菌体结果预测文件】
├── *_prophage.gff3【噬菌体结果文件,gff格式】
└── *_prophage.tsv【噬菌体结果位置信息】
整合在宿主基因组上的温和噬菌体的核酸序列称之为前噬菌体(Prophage),能随宿主细菌DNA进行同步复制或分裂传代。
能与细菌DNA发生重组交叉的特异位点的DNA序列称为attB(att源自attachment)。插入序列左边的一个attB记为attL,插入序列右边的一个attB记为attR。
我们使用PhiSpy[16]分析工具,对细菌基因组prokka注释结果中的.gbk格式文件做分析,得到前噬菌体序列分析结果。
详细结果说明见噬菌体分析结果说明。
表5-1-1 噬菌体结果统计表
| sampleID | Prophage number | Contig | Start | Stop |
|---|---|---|---|---|
| C1 | pp_1 | Chr_1 | 508044 | 536596 |
| C1 | pp_2 | Chr_1 | 1688490 | 1720181 |
| C1 | pp_3 | Chr_1 | 2778761 | 2828280 |
| C2 | pp_1 | Chr_1 | 508044 | 536596 |
| C2 | pp_2 | Chr_1 | 1688862 | 1720181 |
| C2 | pp_3 | Chr_1 | 2778761 | 2828280 |
| Z1 | pp_1 | Chr_1 | 2113568 | 2159514 |
- sampleID: 样本ID
- Prophage number: 噬菌体名字
- Contig: contig名字
- Start: 开始位置
- Stop: 结束位置
- 若是无结果,有可能是样本无噬菌体
3.5.2 crispris序列分析
cripris ├── all_sample.crispr.xls [所有样本cripris注释结果] ├── * | ├──Chr_1.gff [CRISPR基因序列文件,gff格式] | ├──*_crisprcas.gff3[CRISPR基因序列文件,gff格式] | ├──*.CRISPR_information.xls[CRISPR基因结果文件] | ├──*.CRISPR_sequence.xls[包含所有检测到的CRISPR基因序列文件,fasta格式] | ├──*.CRISPR_stat.xls[CRISPR 预测结果统计表] | ├── rawCas.fna[包含所有检测到的Cas基因序列文件,fasta格式] | ├── rawCRISPRs.fna[包含所有检测到的CRISPRss基因序列文件,fasta格式] | ├── result.json[包含检测到的CRISPR阵列和Cas基因的主要信息,JSON文件] | └── Visualization[可视化文件夹] | ├── crispr.css[可视化文件夹css文件] | └── index.html[网页文件]
我们使用CRISPRCasFinder[15]分析工具,对unicycler的组装结果assembly.fasta文件做分析,得到CRISPR-Cas序列分析结果。
详细结果说明见CRISPRCas序列注释结果
表5-2-1 cripris结果统计表(all_sample.crispr.xls)
| sampleID | CRISPR-Cas_Num | Total_length(bp) | Average_length(bp) |
|---|---|---|---|
| C1 | 4 | 841 | 210.2 |
| C2 | 4 | 841 | 210.2 |
- sampleID:样本名
- CRISPR-Cas_Num:CRISPR-Cas数目
- Total_length(bp):总长度
- Average_length(bp):平均长度
3.5.3 插入序列分析
├── *.final.assembly.csv [插入序列结果,CSV格式] ├── *.final.assembly.gff [插入序列结果,gff格式] └── *.final.assembly.sum [插入序列统计表格]
插入序列(insertion sequence,IS)是编码转座所需的酶的一种转座子,它的两侧是短反向末端重复序列。转座子插入的靶序列在插入过程中被复制,在转座子两端先形成两个短正向重复序列。正向重复序列(DR,direct repeat)的长度为5-9bp,是任一转座子的特征。IS是细菌染色体和质粒的正常组成成分。标准的大肠杆菌含有任何一种常见的IS,每一种都有不到10份拷贝。当描述插入特定位置的IS如插入入噬菌体时,可以记为λ::IS。IS都能编码自身转座所需的酶。多数IS元件在宿主DNA内有多个插入位点,但也有些在不同程度上偏爱的特定热点。
我们使用digIS[18]分析工具,对细菌基因组做分析,得到IS插入序列预测结果。
详细结果说明见IS插入序列结果说明。
表5-3-1 插入序列统计表格(all_sample.ISfinder.stat.xls)
| sampleID | #seqid | family | nIS | bps | dnaLen | %dna |
|---|---|---|---|---|---|---|
| C1 | Chr_1 | total | 49 | 55948 | 4782682 | 1.17 |
| C1 | Plas_1 | total | 1 | 867 | 91859 | 0.94 |
| C2 | Chr_1 | total | 49 | 55948 | 4782682 | 1.17 |
| C2 | Plas_1 | total | 1 | 867 | 91859 | 0.94 |
| Z1 | Chr_1 | total | 25 | 34191 | 4041531 | 0.85 |
- sampleID: 样本ID
- #seqid:序列编号
- family:类型(汇总)
- nIS: 插入序列数量
- bps:插入序列总长度
- dnaLen:基因组长度
- %dna:插入序列占基因组长度比例
3.5.4 重复序列预测
├── all_sample.repeatmask.stat.xls [所有样本散在重复序列统计表] ├── all_sample.trfrepeat.stat.xls [所有样本串联重复序列统计表] ├──* │ ├──*.final.assembly.fasta.out [repeat 默认输出结果] │ ├──*.final.assembly.fasta.out.gff [repeat 默认输出结果gff格式] │ ├──*.final.assembly.fasta.tbl [散在重复序列预测结果] │ ├──*.Microsatellite.DNA.dat.gff [Microsatellite DNA 预测结果列表] │ ├──*.Minisatellite.DNA.dat.gff[Microsatellite DNA 预测结果列表] │ ├──*.repeatmask.xls [散在重复序列统计表] │ ├──*.trf.dat.gff [串联重复序列预测结果列表] │ └──*.trf.stat [串联重复序列统计表]
重复序列是基因组中不同位置出现的相同或互补性片段,是基因调控网络的组成成分。 根据重复的序列在基因组上的分布,分为散在重复序列、串联重复序列。
串联重复序列(Tandem Repeat,TR),即相邻的、重复两次或多次特定核酸序列模式的重复序列,分为微卫星(MicroSatellites),1-6个碱基为一个重复单元的简单重复序列(simple sequence repeats),以及10-60个碱基的长序列为一个重复单元的小卫星重复序列(MiniSatellites)。串联重复单元具有种属组成特异性,可作为物种的遗传性状,做进化关系的研究。
散在重复序列主要是转座子序列(transposable elements, TEs)。按长度分为短分散重复序列(Short Interspersed Nuclear Elements,SINEs)以及长散在重复序列(Long Interspersed Nuclear Elements,LINEs),其中长散在重复序列常具有转座活性。
我们使用trf(Tandem Repeats Finder)[13]分析工具,对unicycler的组装结果assembly.fasta文件做分析,得到串联重复序列分析结果。使用RepeatMasker[14]分析工具,得到散在重复序列分析结果。
详细结果说明见重复序列分析结果说明
表5-4-1 串联重复序列统计表格(all_sample.trfrepeat.stat.xls)
| sampleID | Type | Number(#) | Repeat Size(bp) | Total Length(bp) |
|---|---|---|---|---|
| C1 | TR | 91 | 6.0~483.0 | 14568.5 |
| C1 | Minisatellite DNA | 69 | 11.0~100.0 | 5351.1 |
| C1 | Microsatellite DNA | 5 | 6.0~10.0 | 220.0 |
| C2 | TR | 91 | 6.0~483.0 | 14568.5 |
| C2 | Minisatellite DNA | 69 | 11.0~100.0 | 5351.1 |
| C2 | Microsatellite DNA | 5 | 6.0~10.0 | 220.0 |
| Z1 | TR | 123 | 4.0~363.0 | 14498.4 |
| Z1 | Minisatellite DNA | 93 | 11.0~98.0 | 7161.1 |
| Z1 | Microsatellite DNA | 15 | 4.0~8.0 | 830.8 |
- sampleID: 样本ID
- Type: 类型
- Number(#): 数量
- Repeat Size(bp): 重复序列长度范围
- Total Length(bp): 各类型总长度
表5-4-2 串联重复序列统计表格(all_sample.repeatmask.stat.xls)
| sampleID | Type | Number | Total Length(bp) | Ingenome(%) | Average Length(bp) |
|---|---|---|---|---|---|
| C1 | SINEs | 13 | 797 | 0.02 | 61.3 |
| C1 | LINEs | 16 | 983 | 0.02 | 61.4 |
| C1 | LTR elements | 2 | 154 | 0.00 | 77.0 |
| C1 | DNA elements | 7 | 541 | 0.01 | 77.3 |
| C1 | Unclassified | 0 | 0 | 0.00 | 0.0 |
| C2 | SINEs | 13 | 797 | 0.02 | 61.3 |
| C2 | LINEs | 16 | 983 | 0.02 | 61.4 |
| C2 | LTR elements | 2 | 154 | 0.00 | 77.0 |
| C2 | DNA elements | 7 | 541 | 0.01 | 77.3 |
| C2 | Unclassified | 0 | 0 | 0.00 | 0.0 |
| Z1 | SINEs | 15 | 1122 | 0.03 | 74.8 |
| Z1 | LINEs | 7 | 556 | 0.01 | 79.4 |
| Z1 | LTR elements | 1 | 97 | 0.00 | 97.0 |
| Z1 | DNA elements | 8 | 426 | 0.01 | 53.2 |
| Z1 | Unclassified | 0 | 0 | 0.00 | 0.0 |
- sampleID: 样本ID
- Type: 类型
- Number: 数量
- Total Length(bp) : 总长度
- Ingenome(%): 在基因间百分比
- Average Length(bp):平均长度
3.5.5 基因岛预测
基因岛(Genomics Islands,GIs)是一些细菌、噬菌体或质粒中通过基因水平转移整合到微生物基因组中的一个基因组区段。一个基因岛能与病原机理、生物体的适应性等多种生物功能相关。通过比较基因组分析手段可研究具有特殊功能的微生物功能的特异性和功能来源。基于序列组成,采用IslandPath-DIOMB软件预测基因岛,通过检测序列中二核苷酸偏向性(phylogentically bias)和移动性基因(mobility genes,如转座酶或整合酶)以判定基因岛以及潜在的水平基因转移。
详细结果说明见基因岛结果说明。
我们使用IslandPath-DIMOB[17]分析工具,对细菌基因组prokka注释结果中的.gbk格式文件做分析,得到基因岛预测结果,然后用R语言绘制基因岛染色体位置示意图。
表5-5-1 基因岛结果统计表(all_sample.crispr.xls)
| sampleID | GI_num | GI_length(bp) | GI_average_length(bp) |
|---|---|---|---|
| C1 | 11 | 315272 | 28661.09 |
| C2 | 11 | 315272 | 28661.09 |
| Z1 | 6 | 247780 | 41296.67 |
- sampleID:样本名
- GI_num:基因岛数目
- GI_length(bp):总长度
- GI_average_length(bp):平均长度
3.6 基因组功能预测
3.6.1 功能注释
General_Gene_Annotation/
├── CAZy
│ ├── *
│ │ ├── *.CAZy.class.stat.txt [CAZy 数据库注释的结果文件]
│ │ ├── *.CAZy.m8.txt[CAZy 数据库进行 BLAST 比对结果 m8 格式]
│ │ ├── *.CAZy.sumInfo.txt[CAZy 数据库注释的结果文件]
│ │ ├── cazy_summed_up_summary.pdf [CAZy 数据库六大分类统计图,PDF 格式]
│ │ └── cazy_summed_up_summary.png[CAZy 数据库六大分类统计图,PNG 格式]
├── eggnog
│ ├── *
│ │ └── *.emapper.annotations[eggnog结果文件]
├── GO
│ ├── *
│ │ ├── *.gene_go.txt [GO 注释结果按基因汇总]
│ │ ├── *.go.txt[GO 注释结果按分类汇总]
│ │ ├── *.pdf[GO 分类统计图,pdf格式]
│ │ └── *.png[GO 分类统计图]
├── KEGG
│ ├── *
│ │ ├── *.kegg.txt [KEGG 注释结果按分类汇总]
│ │ ├── *.pdf[KEGG 分类统计图,pdf格式]
│ │ └── *.png[KEGG分类统计图]
├── nr
│ ├── *
│ │ ├── *.anno.txt [NR 数据库注释的结果文件]
│ │ ├── *.nr.m8.txt[NR 数据库进行 BLAST 比对结果]
│ │ ├── *.nr.species.anno[NR 数据库物种注释的结果]
│ │ ├── *.nr.species.anno.pdf[NR 数据库物种注释统计图,pdf格式]
│ │ └── *.nr.species.anno.png[NR 数据库物种注释统计图]
├── PFAM
│ ├── *
│ │ ├── *.pfam.gene.tsv [Pfam 注释结果按分类汇总]
│ │ ├── *.pfam.tsv[pfam结果文件]
│ │ └── *.pfam.merge.tsv[Pfam 注释结果按基因汇总]
├── swissProt
│ ├── *
│ │ ├── *.swissProt.m8.txt[Swiss-Prot 数据库进行 BLAST 比对结果]
│ │ └── *.swissProt.mergeInfo.txt[Swiss-Prot 数据库注释的结果文件]
└── TCDB
└── *
├── *.TCDB.anno.tsv[TCDB结果注释文件]
├── tcdb_summed_up_summary.pdf[TCDB 一级分类统计图]
├── tcdb_summed_up_summary.png[TCDB 一级分类统计图]
├── *.TCDB.family.catalog.tsv[TCDB 三级家族分类统计列表]
├── *.TCDB.m8.txt[ TCDB 数据库进行 BLAST 比对结果]
└── *.TCDB.sumInfo.txt[ TCDB 一级分类统计列表]
3.6.1.1 GO数据库注释
GO(Gene Onotology),是生物学领域公认的,在分子和细胞层面的英文描述词条的参考规范。比如一个蛋白具有某种功能,尽管这是一种具体的功能,但是不同的人可能会有不同的描述,此时如果大家都采用GO里面的规范词条去描述,那么就不会出现很多偏门或者杂乱的描述词汇。GO促进了人们对生物学知识的交流和理解。
GO数据库是基因本体联合会(Gene Onotology Consortium)所建立的数据库,旨在建立一个适用于各种物种的,对基因和蛋白质功能进行限定和描述的,并能随着研究不断深入而更新的语言词汇标准。
GO提供了一系列的词条(terms),用于描绘基因(基因产物)的特点,这些词条分为3大类:
(1) 细胞学组件(cellular component),用于描述亚细胞结构、位置和大分子复合物,例如外部封装结构(external encapsulating structure)等。
(2) 分子功能(molecular function),用于描述基因(基因产物)的功能,比如蛋白质结合转录因子活性(protein binding transcription factor activity)。
(3) 生物学过程(biological process),指的是分子功能的有序组合以实现更复杂的生物功能,例如树突状细胞的抗原处理和递呈(dendritic cell antigen processing and presentation)。
使用emapper[29]注释工具,将细菌基因组prokka注释结果中的.faa文件的基因蛋白序列作为query查询序列,到eggnog蛋白数据库做比对搜索,获得GO注释信息,用R语言做分类统计。
详细结果说明见GO数据库注释结果说明。
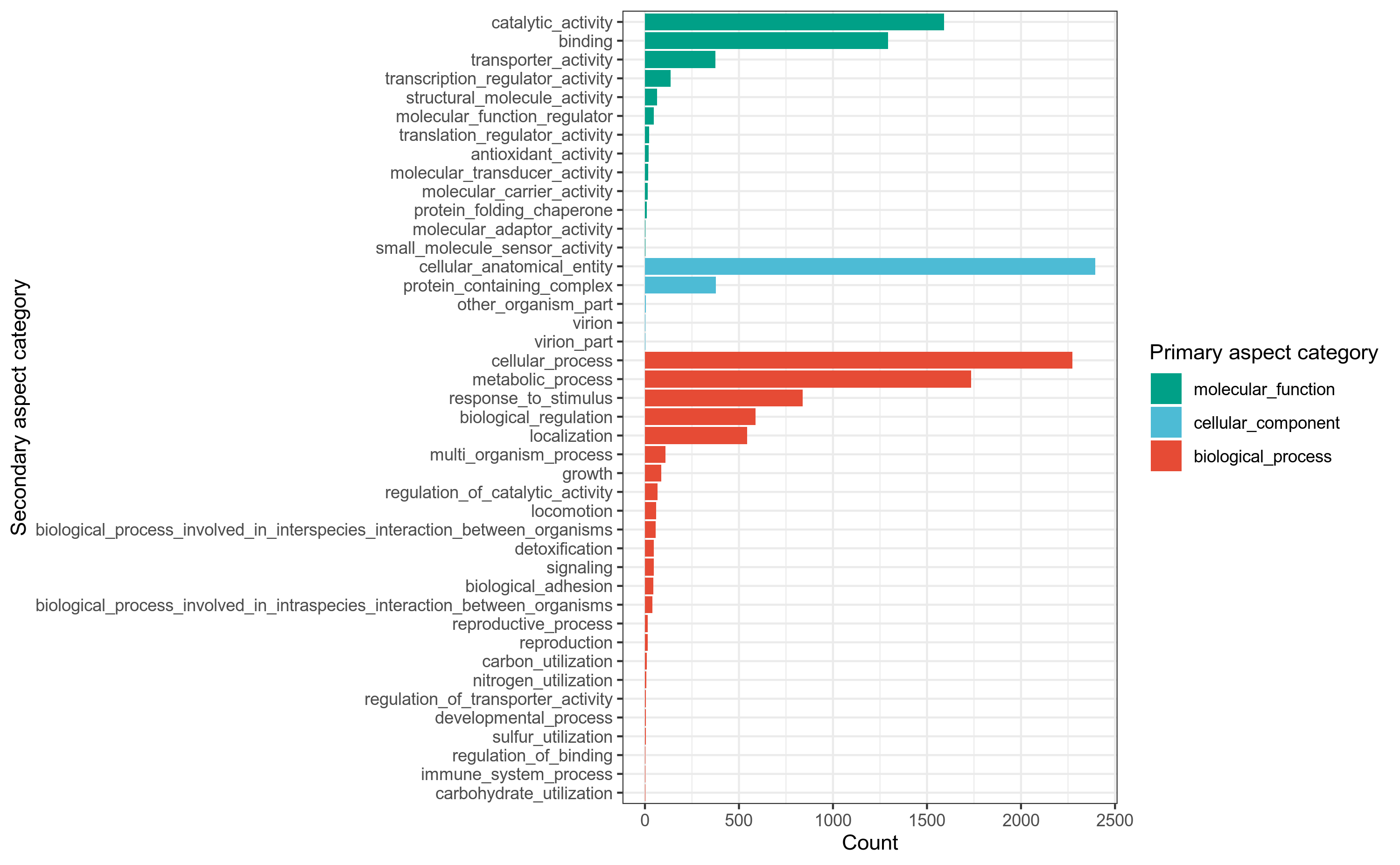
图6-1-1 GO注释分类统计图
3.6.1.2 KEGG数据库注释
KEGG数据库于 1995 年由 Kanehisa Laboratories 推出 0.1 版,目前发展为一个综合性数据库,其中最核心的为 KEGG PATHWAY 和 KEGG ORTHOLOGY 数据库。在 KEGG ORTHOLOGY 数据库中,将行使相同功能的基因聚在一起,称为 Ortholog Groups (KO entries),每个 KO 包含多个基因信息,并在一至多个 pathway 中发挥作用。
在 KEGG PATHWAY 数据库中,将生物代谢通路划分为以下6类:
(1) 细胞过程(Cellular Processes)
(2) 环境信息处理(Environmental Information Processing)
(3) 遗传信息处理(Genetic Information Processing)
(4) 人类疾病(Human Diseases)
(5) 新陈代谢(Metabolism)
(6) 生物体系统(Organismal Systems)
我们使用emapper注释工具,将细菌基因组prokka注释结果中的.faa文件的基因蛋白序列作为query查询序列,到eggnog蛋白数据库做比对搜索,获得KEGG_Pathway_map_id注释信息,用R语言做分类统计。 详细结果说明见KEGG数据库注释结果说明。

图6-1-2 KEGG pathway分类统计图
3.6.1.3 CAZy数据库注释
碳水化合物活性酶(CAZy)数据库,录入的是能降解、修饰或者生成糖苷键的酶的功能结构域(或称模块)的信息。
CAZy数据库[31]收录了碳水化合物活性酶的两种常见模块的数据信息:
(1)具有催化活性的模块,分为5类
GH,Glycoside Hydrolases,糖苷水解酶。
GT,Glycosyl Transferases,糖基转移酶。
PL,Polysaccharide Lyases,多糖裂解酶。
CE,Carbohydrate Esterases,碳水化合物酯酶。
AA,Auxiliary Activities,辅助活性模块(一般是氧化还原酶,跟其它的碳水化合物活性酶共同发生作用)。
(2)结合在催化活性模块之上的其它模块,现有1类
CBM,Carbohydrate-Binding Modules,与碳水化合物发生结合作用的模块,一般只起到结合作用,而没有催化作用。
我们使用蛋白序列比对工具diamond,将prokka注释得到的.faa文件中的蛋白序列作为查询序列,到CAZy数据库做比对搜索,获得CAZy数据库注释信息,然后用R语言对注释结果做分类统计。
详细结果说明见CAZy数据库注释结果说明。
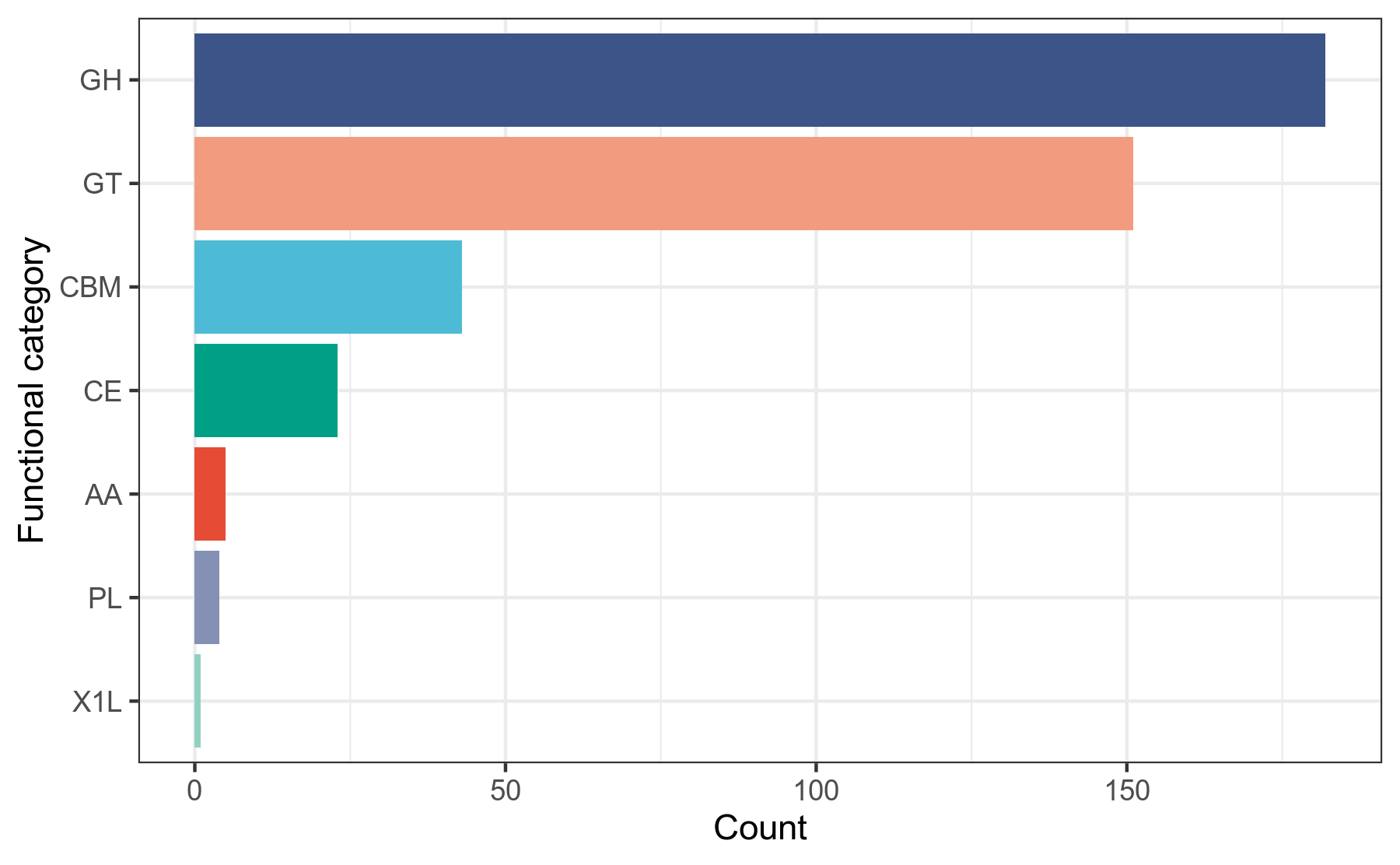
图6-1-3 CAZy数据库注释功能分类统计图
3.6.1.4 SWISS-PROT数据库注释
SWISS-PROT是经过注释的蛋白质序列数据库,由欧洲生物信息学研究所(EBI)维护。数据库由蛋白质序列条目构成,每个条目包含蛋白质序列、引用文献信息、分类学信息、注释等,注释中包括蛋白质的功能、转录后修饰、特殊位点和区域、二级结构、四级结构、与其它序列的相似性、序列残缺与疾病的关系、序列变异体和冲突等信息。SWISS-PROT中尽可能减少了冗余序列,并与其它30多个数据建立了交叉引用,其中包括核酸序列库、蛋白质序列库和蛋白质结构库等。
我们使用diamond蛋白序列比对工具,将细菌基因组prokka注释结果中的.faa文件的基因蛋白序列作为query查询序列,到Swiss-Prot蛋白数据库做比对搜索,得到细菌的Swiss-Prot蛋白注释信息,生成表格文件。
详细结果说明见SWISS-PROT数据库注释结果说明。
3.6.1.5 TCDB数据库注释
TCDB数据库[32](Transporter Classification DataBase)是对转运蛋白进行分类的一个数据库。类似于对酶进行分类的EC系统,TCDB对于每一个转运蛋白家族,提供了一个TC Nmuber, TC Number 由小数点分隔的5段数字或者字母构成。每一段的数字或字母代表某一个层级的分类,第一级分类包括7个大类。目前TCDB提供了超过800个转运蛋白家族, 包含10000多条唯一的蛋白质序列和10000多篇文献。
我们使用diamond比对工具,将细菌基因组prokka注释结果中的.faa文件的基因蛋白序列作为query查询序列,到TCDB数据库做比对搜索,得到细菌的转运蛋白分类注释信息,然后用R语言做分类统计。 详细结果说明见TCDB数据库注释结果说明。

图6-1-4 TCDB转运蛋白分类注释统计图
3.6.1.6 PFAM数据库注释
Pfam数据库[30]可以理解为是Protein family蛋白质家族的英文单词的缩写。该数据库主要提供蛋白质结构域家族的分类信息,被广泛用于查询蛋白质结构域注释信息及其多序列比对信息。在该数据库中,每个蛋白结构域家族由多序列比对和HMMs(hidden Markovmodels,隐马尔可夫模型)所组成。Pfam-A根据最新的UniProtKB蛋白序列数据库所构建而成,是人工注释和检查的蛋白结构域信息数据库,可信度较高。pfam_scan是Pfam官网提供的工具软件,用来分析蛋白序列具有哪些结构域。
我们使用pfam_scan分析工具,以Pfam-A数据库作为参考数据库,对细菌基因组prokka注释结果中的.faa文件的基因蛋白序列做注释,得到蛋白质结构域家族注释信息,生成表格文件。
详细结果说明见PFAM数据库注释结果说明。
3.6.1.7 nr数据库注释
NR全称为Non-Redundant Protein Database,是一个非冗余的蛋白质数据库,由NCBI创建并维护,其特点在于内容比较全面,同时注释结果中会包含有物种信息,可作物种分类用。
详细结果说明见nr数据库注释结果说明。
根据基因注释到的物种情况,统计注释到的物种及基因数目,其统计结果如下图:
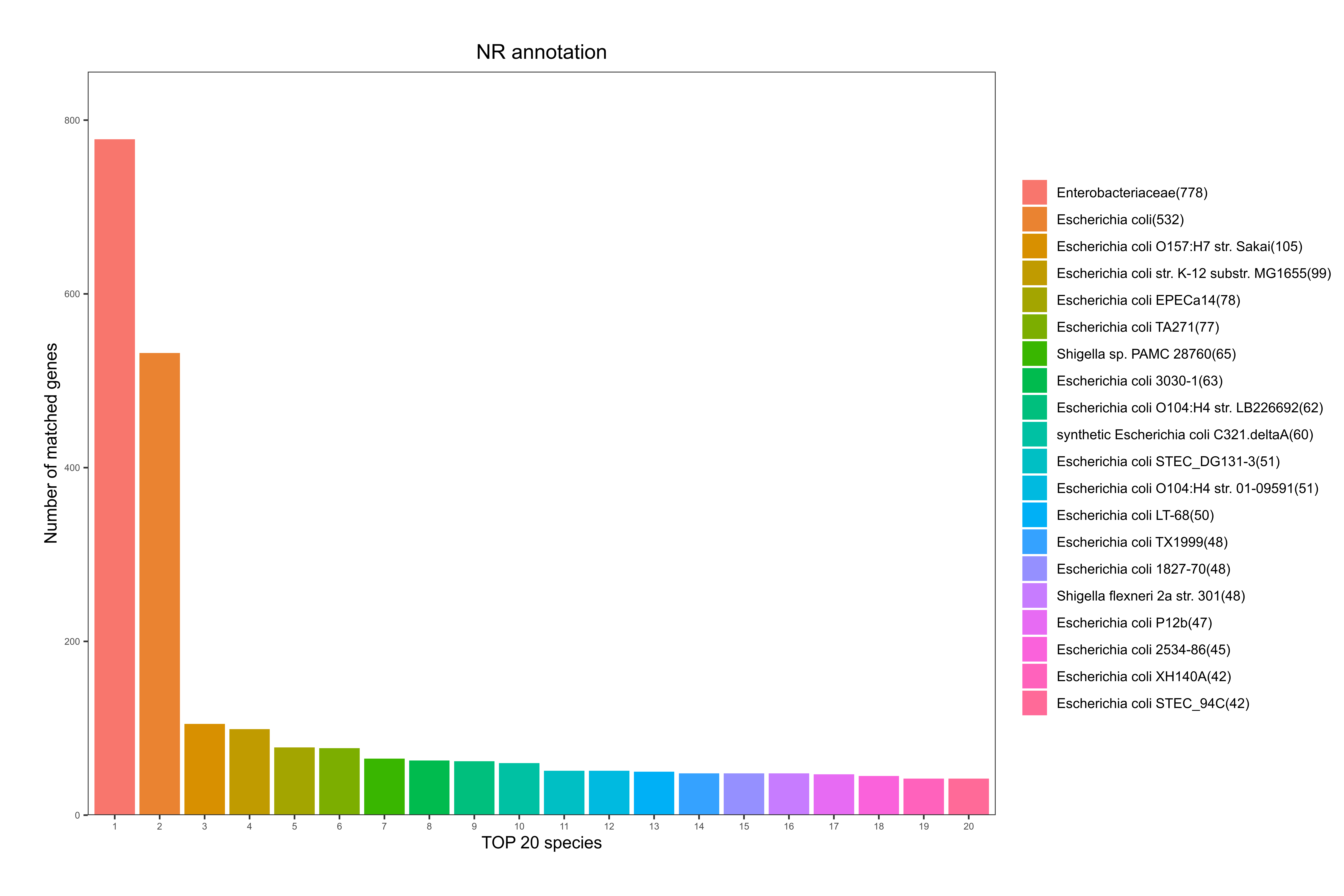
图6-1-5 NR 数据库物种注释统计图
3.6.2 致病系统分析
pathogenicity/
├── CARD
│ ├── *
│ │ └── *.CARD.txt[CARD结果文件]
├── PHI
│ ├── *
│ │ ├── *.phi.anno.txt[PHI数据库注释的结果文件]
│ │ ├── *.phi.filter.m8.txt[PHI 数据库进行 BLAST 比对结果]
│ │ ├── *.phi.sumInfo.txt[PHI 数据库分类统计结果]
│ │ ├── phi_summed_up_summary.pdf[PHI数据库分类统计图]
│ │ └── phi_summed_up_summary.png[PHI数据库分类统计图]
└── VFDB
└── *
├── *.VFDB.anno.txt[VFDB 数据库注释的结果文件]
└── *.VFDB.filter.m8.txt[VFDB 数据库进行 BLAST 比对结果]
3.6.2.1 VFDB数据库注释
毒力因子指由细菌,病毒,真菌等产生的带有侵袭力和毒素等毒力性质的分子,主要用于微生物感染宿主时,通过抑制或逃避宿主的免疫反应等出入宿主组织细胞,并从宿主获得营养及自身增殖生长的目的。毒力因子可编码在可移动遗传元件(比如质粒、基因岛、噬菌体等)上并进行水平基因转移(传播),使无害细菌变成危险的病原菌,所以在鉴定毒力因子时一般会考虑基因岛、分泌蛋白等。VFDB数据库由中国医学科学院研发,收集整理了24个属100多种重要医学病原菌已知毒力因子的组成、结构、功能、致病机理、毒力岛、序列和基因组信息等内容,被广泛应用于毒力因子基因鉴定。
我们使用diamond[24]蛋白序列比对工具,将细菌基因组prokka注释结果中的.faa文件的基因蛋白序列作为query查询序列,到VFDB[26]细菌毒力因子数据库做比对搜索,得到细菌的毒力因子注释信息。
详细结果说明见VFDB数据库注释结果说明。
提示:VFDB官网发布了网页在线分析工具VFanalyzer,推荐用户登录http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi 网站,上传prokka分析结果文件夹中的.gbk注释文件,提交在线分析任务,可以获得关于病原细菌毒力因子更丰富的分析结果。
3.6.2.2 CARD数据库注释
微生物抗生素耐药性问题,是在人类为抑制病原微生物生长繁殖,而高频次、大剂量使用抗生素的背景下凸显出来的。微生物通过自身基因突变,或者在环境中由于基因水平转移,得到的这些(突变的)基因,使药物作用的靶位发生变异或药物不能正常地发挥作用,从而获得对特定抗生素的抗性。
CARD(Comprehensive Antibiotic Resistance Database): 综合性抗生素抗性数据库,是目前使用最广泛的抗性基因数据库之一,目前包括约4000个抗性基因分类。
我们使用card_rgi分析工具,以CARD[27]数据库作为参考数据库,对unicycler的组装结果assembly.fasta文件,做细菌的抗生素抗性基因注释,找出基因组上的抗性基因,生成注释结果表格文件。
详细结果说明见CARD数据库注释结果说明。
3.6.2.3 PHI数据库注释
病原-宿主相互作用,是指微生物在侵染宿主的过程中,跟宿主会有一系列的相互作用,比如宿主需要探测到微生物的某些产物,宿主才能激活正常的免疫反应。那么在这个互作的过程中,微生物的某些基因就发挥着重要的作用。
PHI-base(Pathogen Host Interactions),病原-宿主相互作用数据库。该数据库收录有,微生物中的、能影响病原-宿主相互作用的基因的信息。
其中,我们重点关注,PHI-base收集整理的,在已发表的研究论文中,人为地对微生物的影响病原-宿主相互作用的基因,做基因突变后,造成的微生物菌株的表型信息。
我们使用diamond蛋白序列比对工具,将细菌基因组prokka注释结果中的.faa文件的基因蛋白序列作为query查询序列,到PHI[25]数据库做比对搜索,得到在病原-宿主相互作用中发挥作用的基因,在人为突变后的菌株表型的相关信息,用R语言做分类统计。
详细结果说明见PHI数据库注释结果说明。
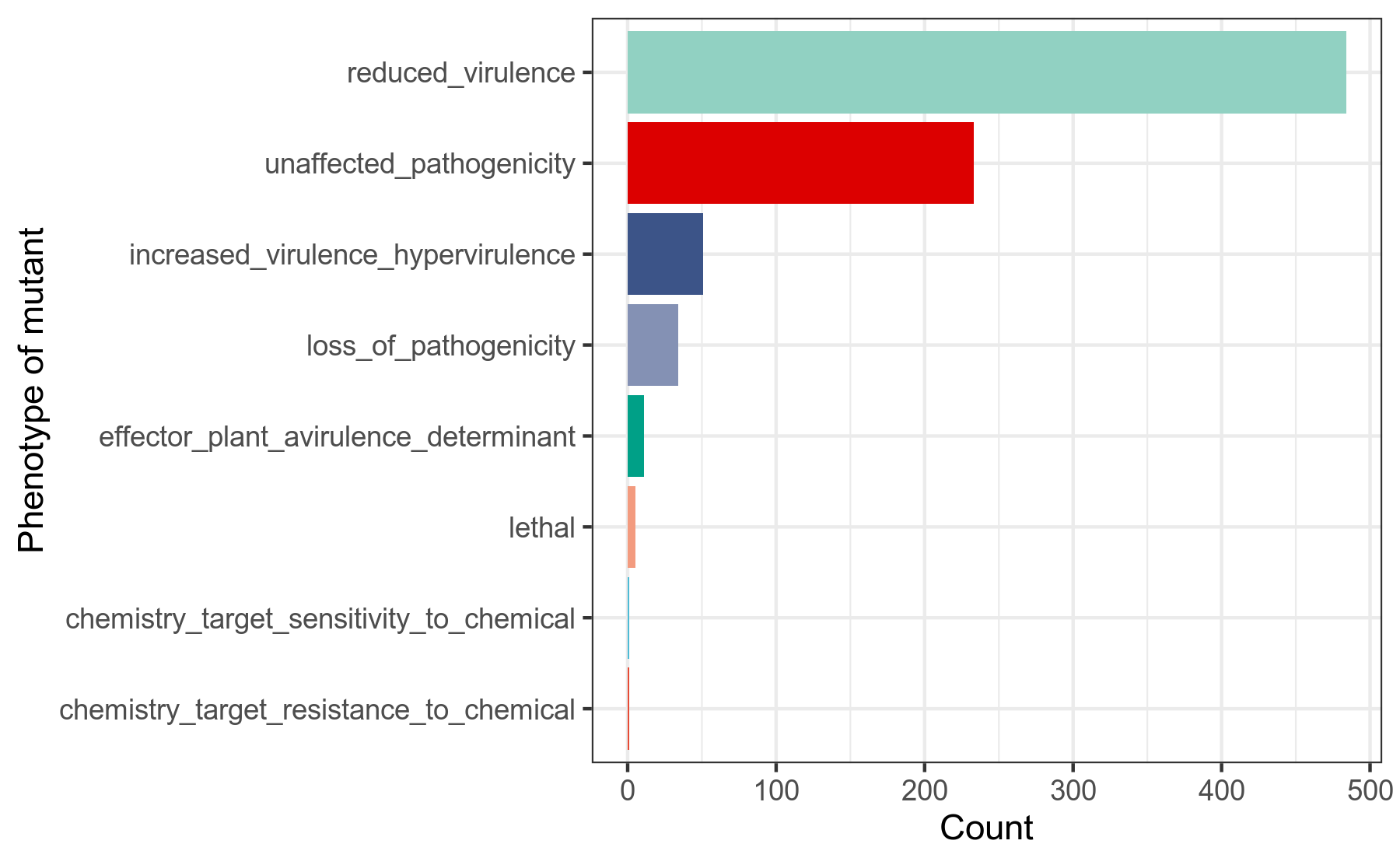
图6-2-1 PHI注释-影响病原宿主互作的基因突变后表型的分类统计图
3.6.3 次级代谢基因簇和效应子预测
metabolism/
├── antiSMASH
│ ├── *
│ │ ├── *.antismash.bed [次级代谢基因簇bed格式结果]
│ │ ├── *.antismash.tsv[次级代谢基因簇列表]
│ │ ├── *.cluster.stat.png[次级代谢基因簇及相应基因的数量统计图,PNG 格式]
│ │ ├── *.cluster.stat.tsv[次级代谢基因簇及基因数量统计]
│ │ └── svg[与已知基因簇比对结果展示图]
│ │ └── clusterblast_*_all.svg [*代表基因簇数字编号]
├── Secretory_Protein
│ ├── all_sample.secretory.stat.xls[所有样本的统计结果]
│ ├── *
│ │ ├── *.membrane.faa[仅包含含有信号肽的序列信息]
│ │ ├── *.region_output.gff3[鉴定为跨膜蛋白的氨基酸序列]
│ │ ├── *.secretory.faa[鉴定为分泌蛋白的氨基酸序列 ]
│ │ ├── *.signalp.output.gff3[包含所有的信号肽区域信息]
│ │ ├── *.sigseq [所有信号肽的氨基酸序列]
│ │ ├── *.tmhmm.txt[跨膜结构预测结果]
│ │ └── Statistics.txt[统计结果]
├── T3SS
│ ├── all_sample.t3ss.stat.xls [所有样本T3SS汇总结果]
│ ├── *
│ │ ├── *.isnot_secretion.faa[不是T3SS分泌蛋白的序列,fasta格式]
│ │ ├── *.is_secretion.faa[T3SS分泌蛋白的序列,fasta格式]
│ │ ├── *.secretion.sum.xlsEffectiveT3 软件预测结果统计]
│ │ └── *.t3ss.txt[EffectiveT3 软件预测结果文件]
└── TNSS
├── all_sample.tnss.stat.xls [所有样本tnss汇总结果]
├── *
│ ├── *.tnss.reslut.txt[结果文件]
│ └── *.tnss.sum.txt [TNSS汇总结果]
3.6.3.1次级代谢物合成基因簇分析
"anti-biotics and Secondary Metabolite Analysis SHell — antiSMASH" 意为抗生素和次级代谢物的分析流程软件。AntiSMASH是用于寻找次级代谢物合成基因簇的最流行的工具之一。AntiSMASH数据库包含约6,200个完整细菌基因组和18,576个细菌草图基因组的注释。
我们使用antiSMASH[23]分析工具,以antiSMASH数据库作为参考数据库,对unicycler的组装结果assembly.fasta文件做分析,得到次级代谢物合成基因簇分析结果。
详细结果说明见次级代谢物合成基因簇结果说明。
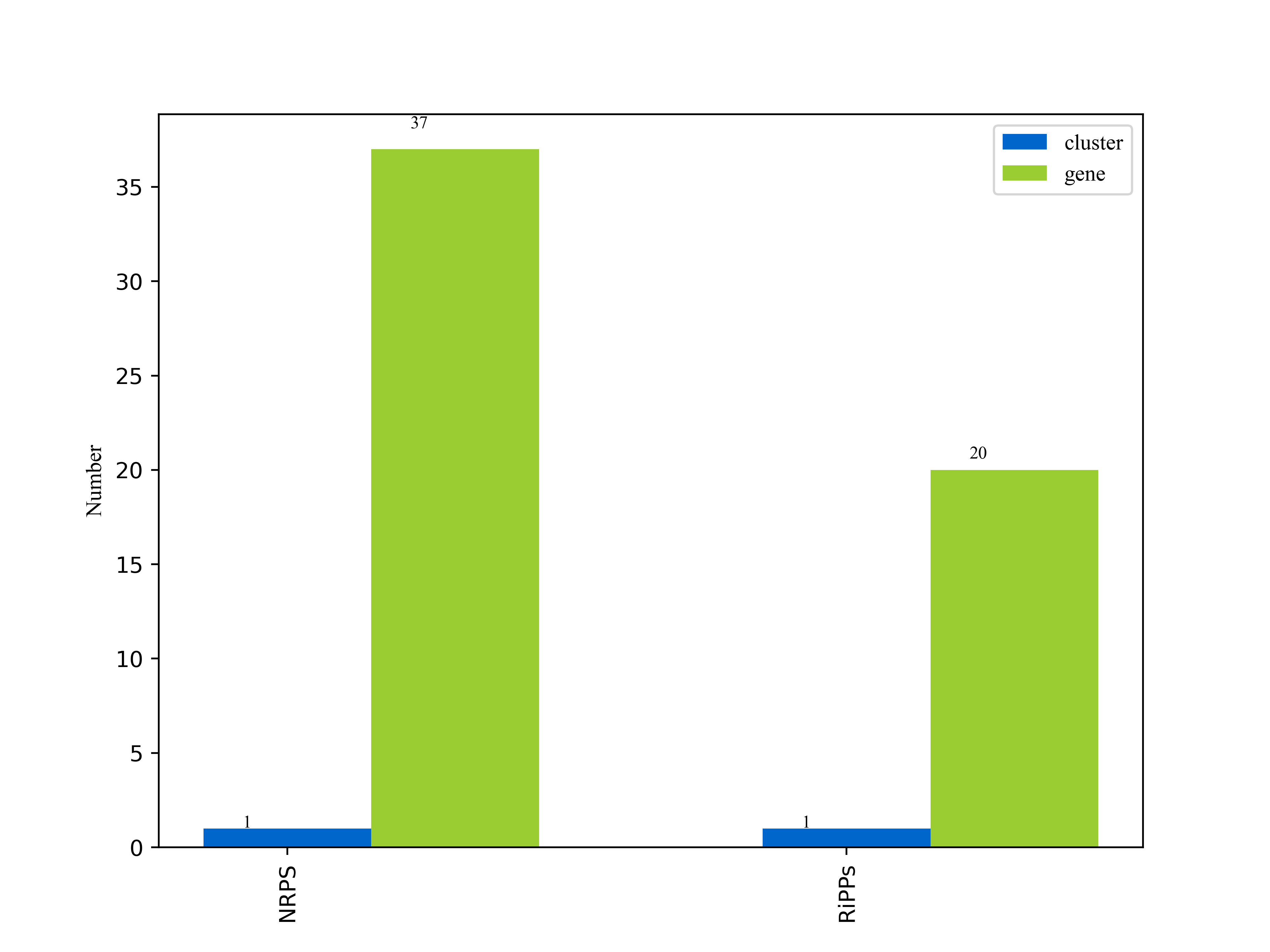
图6-3-1次级代谢物合成基因簇报告
3.6.3.2蛋白信号肽和蛋白跨膜区预测分析
信号肽是蛋白质N-末端一段编码长度为5-30的疏水性氨基酸序列,用于引导新合成蛋白质向通路转移的短肽链。信号肽存在于分泌蛋白、跨膜蛋白和真核生物细胞器内的蛋白中。信号肽指引蛋白质转移的方式有两种:(1)常规的分泌(Sec/secretory)通路;(2)双精氨酸转移(Tat/twin-arginine)通路。前者存在于原核生物蛋白质转移到质膜过程中,以及真核生物蛋白质转移到内质网膜的过程中。后者存在于细菌、古菌、叶绿体和线粒体中,信号肽序列较长、疏水性较弱且尾部区含有两个连续精氨酸。相比于前者转运非折叠蛋白质,后者能转运折叠蛋白质跨越双层脂质膜。
信号肽指引蛋白质转运后,将由信号肽酶进行切除。信号肽酶有三种:(1)一型信号肽酶(SPaseI);(2)二型信号肽酶(SPaseII);(3)三型信号肽酶(SPaseIII)。大部分信号肽由SPaseI进行移除,SPaseI存在古菌、细菌和真核生物中,且在真核生物的内质网膜上仅存在一型信号肽酶。细菌和古菌脂蛋白的信号肽C端含有一段称为 lipobox 的保守区域,由SPaseII切除其信号肽,且lipobox紧邻切除位点(CS/Cleavage Site)的氨基酸是半胱氨酸,这和锚定到膜的功能是相关的。细菌的四型菌毛蛋白信号肽由SPaseIII进行切除。此外:分泌通路(Sec)相关信号肽能由SPaseI、SPaseII和SPaseIII切除,但是双精氨酸转移(Tat)通路相关信号肽仅由 SPaseI和SPaseII切除。
SignalP[19]是最为常用的信号肽分析软件,常用于分泌蛋白预测的第一步。SignalP版本更新到了第六版,结合深度神经网络(deep neural network)、条件随机场分类(Conditional random field classification)和迁移学习(transfer learning)方法,能对信号肽进行更准确的预测。
我们使用SignalP6.0分析工具,对细菌基因组prokka注释结果中的.faa文件的基因蛋白序列做分析,得到蛋白的信号肽预测结果。
蛋白跨膜区即蛋白质序列中跨越细胞膜的区域,通常为α-螺旋结构,约20~25个氨基酸残基,构成跨膜区蛋白的氨基酸大部分是疏水性氨基酸。蛋白质含有跨膜区,提示它可能作为膜受体起作用,也可能是定位在定位在膜上的膜锚定蛋白或离子通道蛋白。TMHMM是最常见的跨膜区分析软件之一,它基于马尔可夫模型,综合跨膜区疏水性、电荷偏倚、螺旋长度和膜蛋白拓扑学等性质,可对跨膜区及膜内外取进行整体预测。
我们使用TMHMM[20]分析工具,对细菌基因组prokka注释结果中的.faa文件的基因蛋白序列做分析,得到蛋白的跨膜区预测结果。
详细结果说明见次级代谢物合成基因簇结果说明。
表6-3-2 分泌蛋白统计结果表
| sample id | Signal peptide numbers | Secretory protein numbers | Membrane protein numbers |
|---|---|---|---|
| C1 | 548 | 424 | 124 |
| C2 | 548 | 424 | 124 |
| Z1 | 481 | 349 | 132 |
- Strain name: 样本ID
- Secretory protein numbers:分泌蛋白数量
- Signal peptide numbers:信号肽数量
- Membrane protein numbers:跨膜蛋白数量
3.6.3.3 TNSS分析
病原菌通过分泌系统TNSS(type N secretion systems)将该类蛋白分泌至胞外或是宿主细胞中,通过控制免疫应答反应以及细胞衰亡引起病理反应,而其中革兰氏阴性菌的T3SS通常用来从分子水平研究病原菌,感染机制,毒力作用等,是研究得比较多的分泌系统。
对于TNSS系统,通过蛋白序列功能数据库注释结果中,提取分泌系统相关蛋白进行注释。
详细结果说明见TNSS结果说明。
表6-3-3 TNSS结果统计表(all_sample_genomesize_stat.xls)
| sampleID | T1SS num | T2SS num | T3SS num | T4SS num | T5SS num | T6SS num | T7SS num | T8SS num | T9SS num | total num |
|---|---|---|---|---|---|---|---|---|---|---|
| C1 | 0 | 7 | 1 | 2 | 0 | 7 | 0 | 0 | 0 | 17 |
| C2 | 0 | 7 | 1 | 2 | 0 | 7 | 0 | 0 | 0 | 17 |
| Z1 | 4 | 1 | 7 | 0 | 0 | 14 | 0 | 0 | 0 | 26 |
- sampleID: 样本ID
- total num:所有TNSS蛋白的数量
- T*SS:各类型分泌系统相关蛋白的个数。
3.6.3.4 T3SS分析
III 型分泌系统(Type III secretion system,T3SS)主要是革兰氏阴性菌的分泌蛋白分泌到细胞外的运输途径,因此 III 型分泌系统效应蛋白(Type III secretion system Effector protein)与革兰氏阴性致病菌致病机理有关。
使用软件 EffectiveT3[21] 对输入的氨基酸序列进行预测,通过其内部特定的计算模型对每条氨基酸序列进行评分,分值越高,可信度越高,选出评分高于阈值的序列,认为这些序列为 III 型分泌系统效应蛋白。
详细结果说明见T3SS结果说明。
表6-3-4 T3SS结果统计表(all_sample_genomesize_stat.xls)
| sampleID | all_pep_sum_num | true_secret_nums | false_secret_nums |
|---|---|---|---|
| C1 | 4572 | 401 | 4171 |
| C2 | 4572 | 401 | 4171 |
| Z1 | 3651 | 401 | 3250 |
- sampleID: 样本ID
- all_pep_sum_num:所有蛋白的数量
- true_secret_nums:是T3SS分泌蛋白数量
- false_secret_nums:不是T3SS分泌蛋白数量 。
- 若是革兰氏阳性菌,则慎重考虑该结果
3.6.4 基因功能注释汇总
目前提供注释的通用功能数据库主要有GO、KEGG、COG、NR、Pfam、TCDB和Swiss-Prot等。
功能注释基本步骤如下:
1)将预测基因的蛋白序列与各功能数据库进行Diamond 比对(evalue ≤ 1e-5);
2)比对结果过滤:对于每一条序列的比对结果,选取 score 最高的比对结果(默认identity>=40%,coverage>=40%)进行注释。
本项目进行的编码基因的注释结果统计如下图所示:
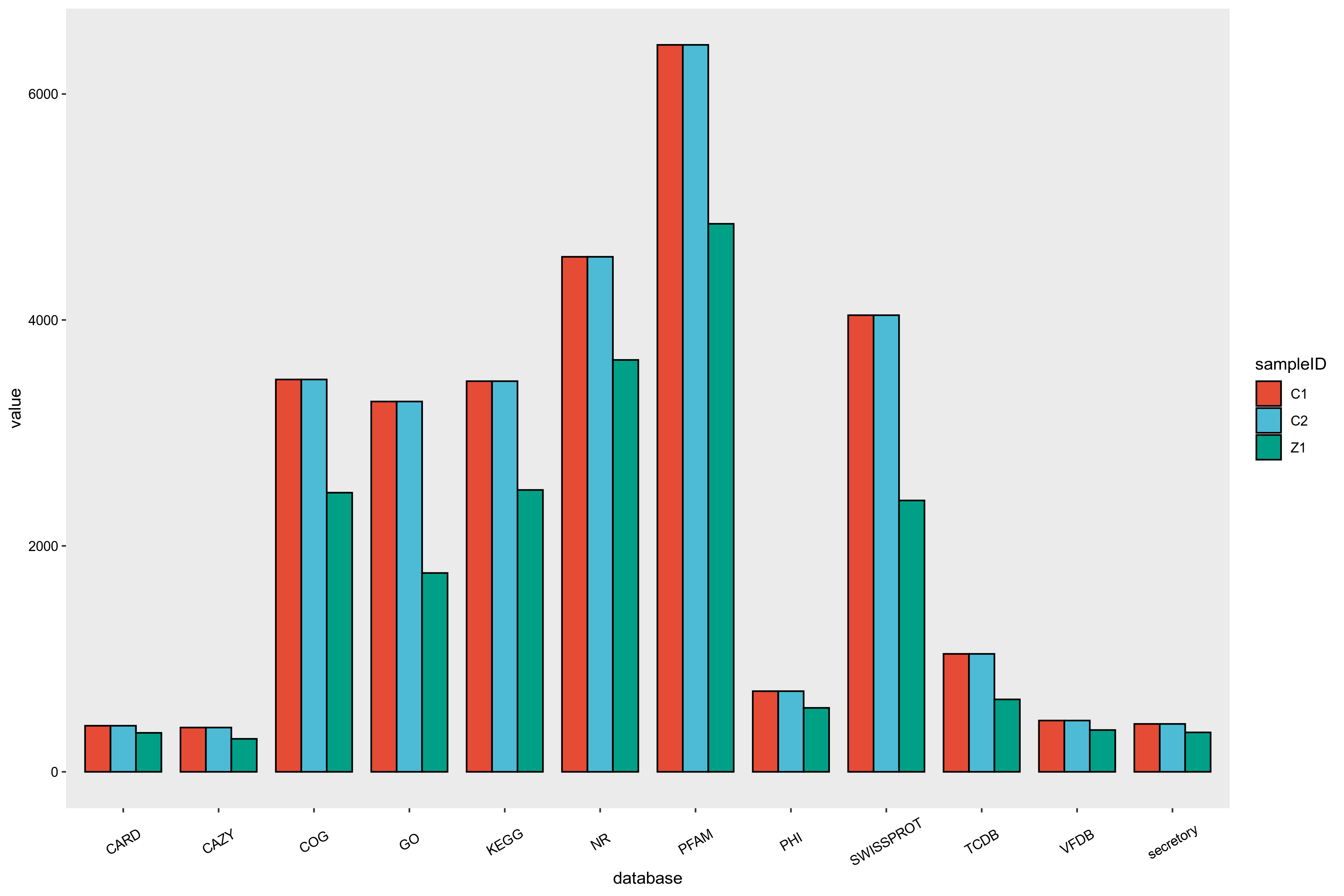
图6-5-1 基因功能分析结果统计图
3.7 基因组可视化
--07.Genome_Drawing/ --*.pdf/ [Circos绘图pdf格式] --*.svg/ [Circos绘图svg格式] --*.png/ [Circos绘图结果图]
Circos是由加拿大生物信息学科学家Martin Krzywinski利用Perl语言开发的一款可以用于描述关系型数据和多维数据的基因组圈图可视化软件。2009年Circos发表在Genome research。Circos不仅能将一个物种的整个基因组呈现在一张图片中,还可以给基因组添加丰富的注释信息,如功能注释信息,差异统计信息等等。
利用细菌基因组prokka[10]蛋白预测信息,功能区注释信息(含正负链、CDS、RNA类型等信息),CAZyme数据库注释结果(GH、GT、CE、PL、CBM、AA),以及GC content、GC skew的统计结果绘制Circos圈图。非常规格式文件请用notepade++或记事本打开。
我们使用Circos[34]软件,分别对细菌基因组的每一条染色体或contig作一个单独的完整圈图,标示基因正负链转录位置信息、RNA位置信息、碳水化合物酶位置信息、基因GC含量信息、基因GC-skew含量等信息。
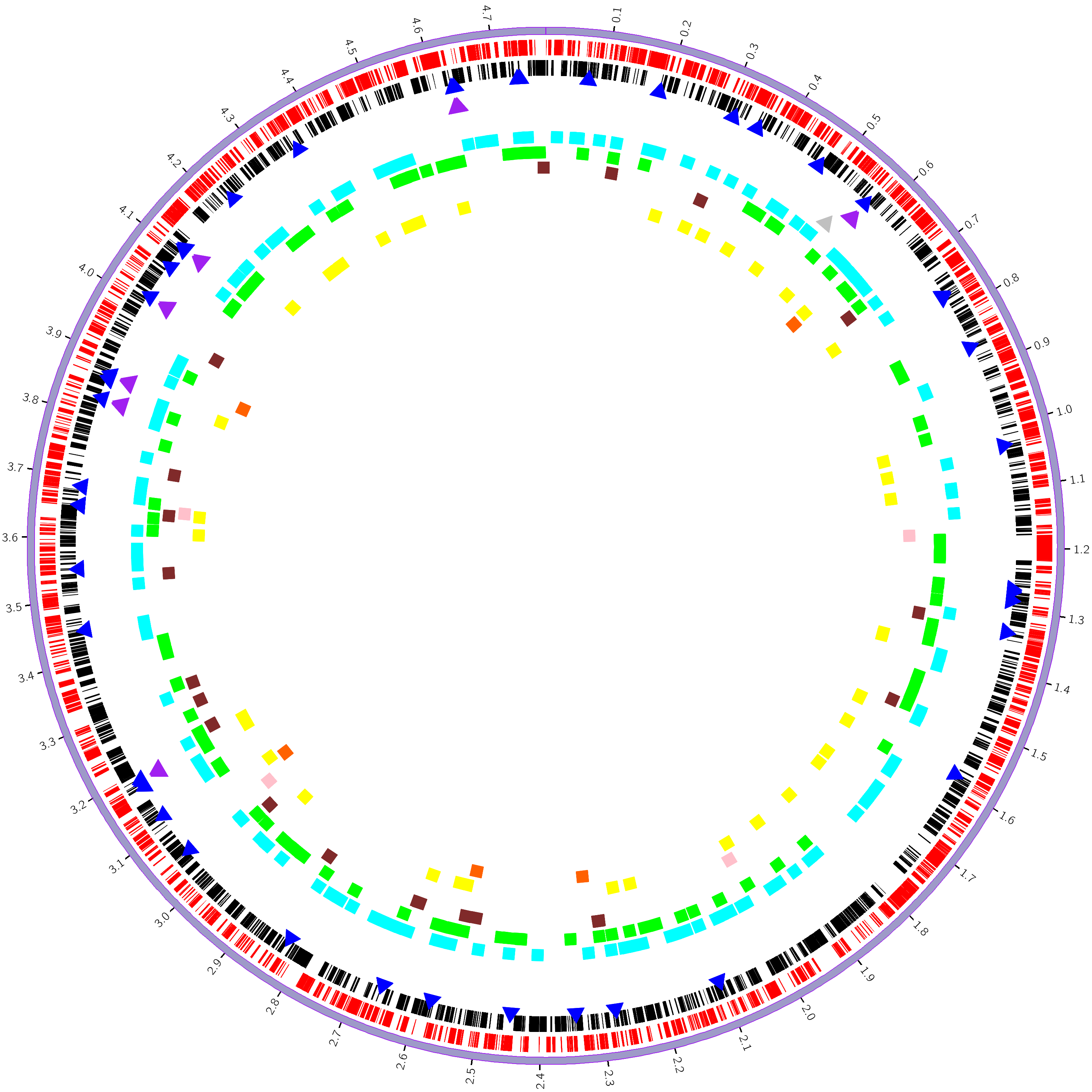
图7-1-1 circos基因组圈图
。这里展示的是对contig1绘制的circos圈图。
从外到内每一圈的含义说明如下:
第一圈:
细菌染色体;
第二圈:
红色高亮表示正链中的CDS区;
黑色高亮表示负链中的CDS区;
第三圈:
蓝色三角表示tRNA;
紫色三角表示rRNA;
灰色三角表示tmRNA;
第四圈:
青色方块表示GH;
绿色方块表示GT;
棕色方块表示CE;
粉红方块表示PL;
黄色方块表示CBM;
橙色方块表示AA;
(方块较大,相邻区域会有重叠,但是完全可以直观的表现不同CAZy的数量差异)
第五圈;
直方图表示基因的GC content,即(G+C)/(A+G+C+T),高度表示GC content的值的大小,宽度表示基因的长度;
红色直方图,方向朝外表示该基因的GC content的值大于基因的总GC content的值;
蓝色直方图,方向朝内表示该基因的GC content的值小于基因的总GC content的值;
第六圈: 直方图表示基因的GC skew(偏移),GC偏移=(G-C)/(G+C)这个式子用来衡量G和C的相对含量,G>C则GC skew的值为正值,G<C则为负值。在大多数细菌基因组中,前导链(leading strand)和滞后链(lagging strand)在碱基组成上有明显的不同——前导链富含G和T,但是滞后链中的A和C则更多一些。打破A=T和C=G的碱基频率发生的偏移,被称之为“AT(AT-skew)”和“GC(GC-skew)”。由于通常GC偏移比AT偏移发生的更明显,所以习惯上更多地只考虑GC偏移。 橙色直方图表示contig的GC skew>0,高度表示绝对值大小; 绿色的直方图表示contig的GC skew<0,高度表示绝对值大小;
四 分析所用软件的版本
| 软件 | 版本 |
|---|---|
| bam2fastq | 1.3.1 |
| fastp | 0.23.1 |
| quast | 2.1.5 |
| unicycler | 2.2.1 |
| bwa | 0.7.17-r1188 |
| Jellyfish | 1 |
| GenomeScope | V1 |
| prokka | 1.14.6 |
| CompareM | 2.2.1 |
| pyani | 2.2.1 |
| Tandem Repeat Finder | 2.1.5 |
| RepeatMasker | v0.12.6 |
| CRISPRCasFinder | v0.12.6 |
| PhiSpy | v0.12.6 |
| IslandPath-DIMOB | v0.12.6 |
| digIS | v1.2 |
| SignalP | v6.0 |
| tmhmm | v2.0 |
| EffectiveT3 | 2.0 |
| MacSyFinder | 2.0 |
| antiSMASH | v6.0 |
| diamond | v2.0.14 |
| PHI | 2022 |
| VFDB | 2022 |
| CARD | 2020 |
| Diamond | v2.0.14 |
| eggNOGmapper | 104.3 |
| pfam | 35.0 |
| CAZy | 2022 |
| tcdb | 2022 |
| emapper | 2.0.1 |
| Circos | 0.69 |
| SMRT Link | v11 |