温馨提示:请使用火狐或者Chrome的网页浏览器来查看报告
真菌精细图分析结题报告
一、 概述
真菌基因组研究,是通过基因组测序和组装获得真菌全基因组序列,并对其进行结构和功能研究的方法。 真菌基因组测序为真菌的研究提供强有力的支撑,可用于预测真菌的重要基因和蛋白以了解其功能和可能机制。
真菌精细图分析对三代测序数据和二代测序数据进行组装,获得真菌全基因组序列,并使用基因组组分分析、 基因功能分析、比较基因组分析、群体进化分析和甲基化分析等分析方法,对真菌基因组相关信息进行全面、详尽地了解。
二、 工作流程概述
2.1 实验流程
2.1.1 文库构建及库检
(1)DNA的提取
采用SDS或STE的方法对样本的基因组DNA进行提取,之后利用琼脂糖凝胶电泳检测DNA的纯度和完整性,利用Qubit进行定量。
(2)Pacbio平台建库及库检
采用SMRT bell TM Template kit(version 1.0)试剂盒构建20K SMRT Bell文库,将经电泳检测合格的DNA样品用Covaris g-TUBE打断成构建文库所需大小的目的片段,经DNA损伤修复及末端修复,使用DNA黏合酶将发卡型接头连接在DNA片段两端,并使用AMpure PB磁珠对DNA片段进行纯化,使用BluePipin片段筛选特定大小的片段,使用AMpure PB磁珠对SMRT Bell文库进行浓度筛选,随后修复DNA损伤,再次使用AMpure PB磁珠对SMRT Bell文库纯化,将构建好的文库经Qubit浓度定量,并利用Agilent 2100检测插入片段大小,最后用PacBio平台进行测序。
(3)Illumina平台建库及库检
经电泳检测合格的DNA样品用Covaris超声波破碎仪随机打断成长度约为350bp的片段。处理完成后的DNA片段,使用NEBNext®Ultra™ DNA Library Prep Kitfor Illumina(NEB, USA)试剂盒,经末端修复、加A尾、加测序接头、纯化、PCR扩增等步骤完成整个文库制备。
文库构建完成后,先使用Qubit 2.0进行初步定量,稀释文库至2ng/ul,随后使用Agilent 2100对文库的插入片段进行检测,insert size符合预期后,使用Q-PCR方法对文库的有效浓度进行准确定量,以保证文库质量。
2.1.2 建库类型
2.1.3 上机测序
库检合格后,把不同文库按照有效浓度及目标下机数据量进行PacBio Sequel和Illumina NovaSeq PE150测序。
2.2 生物信息分析流程
图1 生物信息分析流程图
信息分析分以下几个步骤:
1 原始下机数据处理:此步骤过滤测序质量值低的reads,保留高质量reads,过滤后的数据称为Clean Data;
2 样品组装:进行基因组组装,得到能反映样品基因组基本情况的序列文件,并对组装结果进行评价;
3 基因组组分分析:组装完成后,分析样品基因组的成分,包括编码基因、非编码RNA、重复序列等基因组成分的预测;
4 基因功能分析:针对编码基因序列进行不同数据库的功能注释,包括常用的KEGG、KOG数据库、针对致病性的数据库;
5 比较基因组分析:此步骤从基因组、基因两层面分别比较样品与参考基因组的差异,包括共线性分析、SNP统计与注释、InDel统计与注释、SV统计与注释;
6 群体进化分析:此步骤包括共有基因及特有基因、基因家族分析和群体进化分析等内容;
7 甲基化分析:此步骤对最终的基因组组装结果进行甲基化位点检测和可能的甲基化转移酶识别的核苷酸基序(motif)的预测,包括表观修饰识别、甲基化motif及未甲基化motif在GR/IGR上的分布统计、motif基因注释、COG注释分布图、甲基化圈图等内容。
三、 分析结果
3.1 数据概况
customer_files/01reads_info/
├── 01.01NGS_reads_info
│ ├── ErrorRate.[样本].svg [各样本二代测序数据reads错误率分布图]
│ ├── GCContentDistribution_1.[样本].svg [各样本二代测序数据reads碱基含量分布图]
│ ├── QualityDistribution.[样本].svg [各样本二代测序数据reads测序质量分布图]
│ └── [样本].svg [各样本二代测序数据reads数据过滤统计图]
└── 01.02TGS_reads_info
├── [样本].reads_length.svg [各样本reads长度分布图]
└── read_length_table.txt [所有样本reads长度信息汇总表]
真菌精细图测序数据包含二代测序数据和三代测序数据,以下内容分布对二代测序数据和三代测序数据概况进行展示。
3.1.1 二代测序数据概况
测序获得的原始数据中包含少量带有测序接头或测序质量较低的reads,为保证数据分 析的质量及可靠性,需要对原始数据进行过滤。本分析使用Trimmomatic[1]软件对 测序数据进行过滤,过滤前后各部分reads所占比例均在饼图中呈现。本项目测序数 据过滤情况统计图见结果文件 customer_files/01Trimmomatic。下图为其中一个样本作为示例:

图3-2 测序数据过滤情况
注:
Both Surviving:该部分为过滤后read1和read2均被保留的数据
Forward Only Surviving:该部分为仅在read1中被保留的数据
Reverse Only Surviving:该部分为仅在read2中被保留的数据
Dropped:该部分为因接头或数据质量等原因被丢弃的数据
3.1.3 测序错误率分布
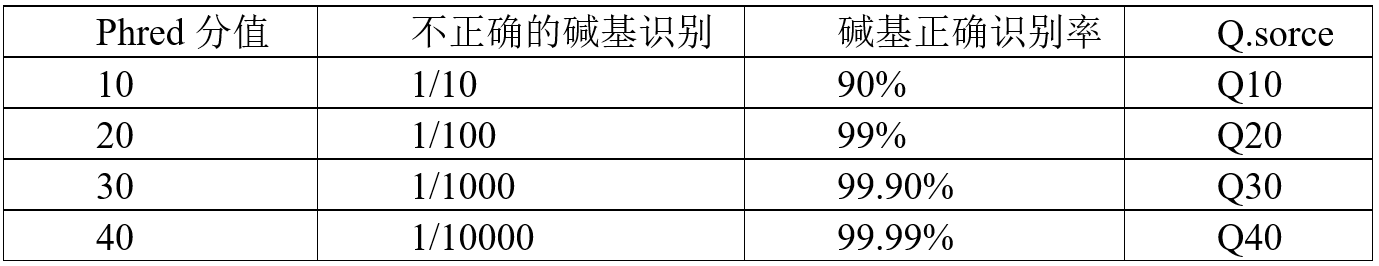
测序过程本身存在机器错误的可能性,测序错误率分布检查可以反映测序数据的质量, 序列信息中每个碱基的测序质量值保存在fastq文件中。如果测序错误率用e表示, Illumina的碱基质量值用Qphred表示,则有:Qphred=-10log10(e)。 Illumina Casava 1.8版本碱基识别与Phred分值之间的简明对应关系见下表。
表3.1 Illumina Casava 1.8版本碱基识别与Phred分值之间的简明对应关系
当前RNA-seq测序技术,测序错误率分布存在以下两个特征。
测序错误率随着测序序列(Sequenced Reads)长度的增加而升高。 这是由测序过程中化学试剂的消耗导致的,为Illumina高通量测序平台所具有的特征。
前6个碱基具有较高的测序错误率,此长度恰好为RNA-seq建库过程中反转录所需的随机引物长度。 前6个碱基测序错误率较高是因为随机引物和RNA模版的不完全结合。 此特征为illumina高通量测序平台的共有特征。
在该部分分析中,若样品80%的测序序列错误率在0.1%以下即为合格。 本项目测序数据的错误分布图见结果文件 customer_files/02QC/02.01ErrorRate/。 下图为其中一个样本作为示例:

图3-3 测序错误率分布
注:
pos:横坐标为reads碱基位置,其中从0-150为read1碱基位置,151-300为read2碱基位置。
errorRatio:纵坐标为碱基错误率。
3.1.4 GC含量分布
核苷酸序列中鸟嘌呤(G)和胞嘧啶(C)所占的比例称为GC含量。 GC含量在物种间存在一定特异性,但由于反转录过程中所使用的6bp随机引物, 会引起前几位碱基在核苷酸组成上有一定偏好性,产生正常波动,随后则趋于稳定。 对于NEB普通建库方法,由于序列的随机性打断和双链互补等原则, 理论上测序读段在每个位置的GC及AT含量应分别相等,且在整个测序过程基本稳定不变, 呈水平线。而对于链特异性建库而言,由于只保留了单链信息, 可能会出现AT分离或GC分离现象。
本项目各个样本的GC含量分布见结果文件 /customer_files/02QC/02.02GCContent。 下图为其中一个样本作为示例:

图3-4 样本GC含量分布
注:
base position:横坐标为reads碱基位置;
base count:纵坐标为该碱基所占比例。
3.1.5测序数据质量分布
测序数据的质量主要分布在 Q30(≥80%)以上,这样才能保证后续分析的正常进行, 根据测序技术的特点,测序片段末端的碱基质量一般会比前端的低。
本项目各个样本测序数据质量分布见结果文件 customer_files/02QC/02.03QualityDistribution。 下图为其中一个样本作为示例:

图3-5 样本测序质量分布
注:
pos:横坐标为碱基位置;
quality:碱基的测序质量值。
3.1.2 三代测序数据概况
测序得到的原始数据会存在一定比例的低质量数据,为了保证后续信息分析结果的准确可靠,首先要对原始数据进行过滤处理,得到有效数据。详细的质控统计信息如下表:
表1 测序数据统计
| samples | number of reads | number of bases | mean read length | N50 |
|---|---|---|---|---|
| sallet_1 | 150000 | 1792442690 | 11949.617933333333 | 11757 |
| sallet_2 | 150000 | 1811985479 | 12079.903193333334 | 17590 |
| sallet_3 | 150000 | 1807866049 | 12052.440326666667 | 12987 |
| sallet_4 | 150000 | 1813879445 | 12092.529633333334 | 6914 |
| sallet_5 | 150000 | 1797219380 | 11981.462533333333 | 13226 |
| sallet_6 | 150000 | 1789891908 | 11932.61272 | 11005 |
说明: samples:样本名称; number of reads:测序数据reads数量; number of bases:测序数据碱基数量; mean read length:测序数据reads平均长度; N50:N50长度。
过滤后得到的有效数据的测序读长分布情况如下图显示:

图2 测序reads长度分布
说明: 横坐标read length为read长度,纵坐标read count为对应read长度的read数量。
3.2 基因组概况
customer_files/02assemble/
├── 02.01Initial_assembly/
│ └── [物种].[样本].contigs.fasta [各样本初步组装contig序列]
├── 02.02GC_depth_plot/
│ └── [物种].[样本].GC_depth_count.svg [各样本GC_depth图]
└── 02.03purged_genome/
├── [物种].[样本].purged.fa [各样本优化后序列]
└── purged_seq_info_count.txt [优化后序列信息汇总]
3.2.1 组装分析
从各样品质控后的有效数据出发,使用canu v2.2软件对reads进行基因组组装,得到能反映样品基因组基本情况的初步的组装结果。
表2 样品基因组初步组装结果统计
| sample ID | Contigs | Max_Length(bp) | N50_Length(bp) | Total_length(bp) |
|---|---|---|---|---|
| sallet_1 | 31 | 5345221 | 2876665 | 34428771 |
| sallet_2 | 32 | 5273289 | 2772635 | 36626721 |
| sallet_3 | 32 | 5371789 | 2788835 | 38838744 |
| sallet_4 | 30 | 5332739 | 2999835 | 36664632 |
| sallet_5 | 31 | 5474477 | 2777645 | 37772531 |
| sallet_6 | 33 | 5537727 | 2225365 | 38887312 |
说明: sample ID:样本名称; Contigs:样本组装contig数量; Max_Length(bp):样本最长contig长度; N50_Length(bp):样本contig N50长度; Total_length(bp):样本组装contig总长度。
3.2.2 组装结果评价
在获得初步组装结果后,使用pbmm2软件将reads比对到组装好的序列上,然后使用samtools软件对reads测序深度进行统计;同时对组装序列的GC含量进行统计,总结组装序列的GC偏向性和重复序列情况,然后使用R脚本对GC含量和测序深度进行可视化展示。
分析结果如下图所示:

图3 样品GC含量与测序深度(Depth)关联分析统计图
说明:
横坐标GC参与组装的read的GC含量,纵坐标depth为参与组装的read的评价测序深度。上方的柱状图为GC含量对应的柱状图,右侧柱状图为测序深度对应的柱状图。
3.2.3 组装结果优化
在获得初步组装结果后,分别使用pilon软件和gcpp软件进行二代测序矫正和三代测序自我矫正。然后使用Purge_dups软件对矫正后序列进行去冗余,从而获得优化后组装序列,即为最终的组装结果。对最终的组装结果统计如下:
表3 最终组装结果信息统计
| sample ID | Contigs | Max_Length(bp) | N50_Length(bp) | Total_length(bp) |
|---|---|---|---|---|
| sallet_1 | 17 | 5251536 | 2762663 | 36266321 |
| sallet_2 | 16 | 5626263 | 2766251 | 32166732 |
| sallet_3 | 18 | 5772636 | 2672783 | 33777382 |
| sallet_4 | 19 | 5626531 | 2888831 | 38872731 |
| sallet_5 | 16 | 5665212 | 2673712 | 33377612 |
| sallet_6 | 17 | 5736641 | 2863721 | 32737841 |
说明: sample ID:样本名称; Contigs:样本组装contig数量; Max_Length(bp):样本最长contig长度; N50_Length(bp):样本contig N50长度; Total_length(bp):样本contig总长度。
3.3 基因组组分分析
customer_files/03genomeComponent/
├── 03.01gene_predict/
│ ├── [物种].[样本].augustus.protein.fa [各样本预测基因对应蛋白质序列]
│ ├── [物种].[样本].augustus.gff [各样本预测基因gff文件]
│ ├── [物种].[样本].coding_gene_length.svg [各样本预测基因长度分布图]
│ ├── [物种].[样本]_coding_gene_length.txt [各样本预测基因长度信息]
│ └── coding_gene_info_count.txt [各样本预测基因长度汇总信息]
├── 03.02repeat_seq/
│ ├── tandem_repeat_count.svg [串联重复统计图]
│ ├── tandem_repeat_count.txt [串联重复统计表]
│ └── [物种].[样本].interspersed_repeat_count.txt [各样本散在重复序列统计表]
└── 03.03ncRNA_predict/
├── ncRNA_count.txt [ncRNA统计表]
├── [物种].[样本].purged.tblout.dealed.table [ncRNA预测结果]
├── [物种].[样本].purged.tRNA.out [tRNA预测结果]
└── [物种].[样本].purged.tRNA.structure [tRNA预测的结构文件]
微生物基因组包含的功能区域非常丰富,除编码基因区域,更有非编码区域实现转录调控、转录后调控、翻译调控、表观遗传调控等功能,部分功能区域还与物种进化的多样性存在关系。 通过多种方法,对编码基因、重复序列、非编码RNA等进行预测,获取目标基因组的组成情况。
3.3.1 编码基因
根据获得的组装序列,我们使用Augustus软件对真菌样本的编码基因进行预测。该软件基于HMM(隐马尔科夫模型)和贝叶斯理论,根据序列信息对其中的编码基因进行预测。基因预测结果统计信息如下表所示:
表4 编码基因预测结果统计表
| sample ID | Genome size | Gene number | Gene total length | Gene average length | Gene length / Genome |
|---|---|---|---|---|---|
| sallet_1 | 33897508 | 12626 | 20626532 | 1682.23 | 0.608497002 |
| sallet_2 | 34332212 | 11233 | 22177631 | 1667.76 | 0.645971515 |
| sallet_3 | 33773642 | 11212 | 26553222 | 1666.21 | 0.786211389 |
| sallet_4 | 33827361 | 12312 | 24343221 | 1677.22 | 0.7196311 |
| sallet_5 | 33737642 | 14321 | 21223426 | 1683.95 | 0.629072595 |
| sallet_6 | 33456212 | 12121 | 24421222 | 1624.25 | 0.729945817 |
说明: Genome size:全基因组总长度; Gene number:预测到的编码基因个数; Gene total length:所有编码基因的总长度; Gene average length:编码基因的平均长度; Gene length / Genome:编码区总长度占全基因组的比例。
绘制基因长度统计图如下:

图4 基因长度统计图
说明:
横坐标gene length为预测的基因长度区间,纵坐标gene number为对应长度区间内的基因数量。
3.3.2 重复序列
重复序列是基因组中不同位置出现的相同或互补性片段,是基因调控网络的组成成分。 根据重复的序列在基因组上的分布,分为散在重复序列、串联重复序列。
散在重复序列又分短分散重复序列(Short interspersed nuclear elements,SINEs)以及长散在重复序列(Longinterspersed nuclear elements,LINEs),其中长散在重复序列常具有转座活性。串联重复序列(Tandem Repeat,TR),即相邻的、重复两次或多次特定核酸序列模式的重复序列。分为Minisatellite DNA(小卫星DNA)和Microsatellite DNA(微卫星DNA)。串联重复单元具有种属组成特异性,可作为物种的遗传性状,进行进化关系的研究。
通过RepeatMasker软件进行散在重复序列预测,TRF(Tandem Repeats Finder)搜寻DNA序列中的串联重复序列。
预测结果如下表所示:
表5 重复序列信息统计
| samples | Minisatellite DNA number | Minisatellite DNA length range | Minisatellite DNA totle length | Minisatellite DNA in genome(%) | Microsatellite DNA number | Microsatellite DNA length range | Microsatellite DNA totle length | Microsatellite DNA in genome(%) | other tandem repeat number | other tandem repeat length range | other tandem repeat totle length | other tandem repeat in genome(%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sallet_1 | 1045 | 29_211 | 64660 | 0.00552517 | 236 | 2_221 | 12442 | 0.000344241 | 568 | 2043_87230 | 222163 | 0.00021113 |
| sallet_2 | 1036 | 21_364 | 64542 | 0.00215551 | 245 | 2_234 | 12032 | 0.000355222 | 518 | 2140_87031 | 226331 | 0.00033214 |
| sallet_3 | 1023 | 22_444 | 65552 | 0.00190721 | 221 | 2_322 | 12212 | 0.000365321 | 535 | 2021_87020 | 226632 | 0.00065321 |
| sallet_4 | 1025 | 26_321 | 67663 | 0.00192121 | 235 | 2_222 | 12332 | 0.000213451 | 558 | 2022_87010 | 227631 | 0.00035762 |
| sallet_5 | 1036 | 26_432 | 64213 | 0.00177632 | 312 | 2_342 | 12432 | 0.000322455 | 521 | 2034_87110 | 228732 | 0.00021763 |
| sallet_6 | 1047 | 29_421 | 64221 | 0.00214511 | 332 | 2_231 | 12541 | 0.000213456 | 451 | 2040_87030 | 229221 | 0.00035521 |
说明: samples:样本名称; Minisatellite DNA number:Minisatellite DNA数量; Minisatellite DNA length range:Minisatellite DNA长度范围; Minisatellite DNA totle length:Minisatellite DNA总长度; Minisatellite DNA in genome(%):Minisatellite DNA长度占基因组长度百分比; Microsatellite DNA number:Microsatellite DNA数量; Microsatellite DNA length range:Microsatellite DNA长度范围; Microsatellite DNA totle length:Microsatellite DNA总长度; Microsatellite DNA in genome(%):Microsatellite DNA长度占基因组长度的百分比; other tandem repeat number:other tandem repeat数量; other tandem repeat length range:other tandem repeat长度范围; other tandem repeat totle length:other tandem repeat总长度; other tandem repeat in genome(%):other tandem repeat长度占基因组长度的百分比。
重复序列统计可视化如下:
图5 重复序列统计图
说明:
横坐标samples为不同的样本,纵坐标count为各个样本中各类重复序列的数量。
3.3.3 非编码RNA
非编码RNA(ncRNA)是一类执行多种生物学功能的RNA分子,其本身并不携带翻译为蛋白质的信息,直接在RNA水平对生命活动发挥作用。对于微生物而言,研究较为普遍的包括sRNA、rRNA、tRNA。
tRNA:转运RNA(Transfer RNA),又称传送核糖核酸、转移核糖核酸,通常简称为tRNA,是一种由76-90个核苷酸所组成的RNA,其3'端可以在氨酰-tRNA合成酶催化之下,接附特定种类的氨基酸。转译的过程中,tRNA可借由自身的反密码子识别mRNA上的密码子,将该密码子对应的氨基酸转运至核糖体合成中的多肽链上。本分析中通过tRNAscan-SE软件对tRNA进行预测。
rRNA:即核糖体RNA,rRNA在相邻物种中高度保守。rRNA的预测方法有两种,一是通过与近缘参考序列的rRNA库比对找到rRNA,二是用rRNAmmer软件预测rRNA。
sRNA:小RNA,首先进行Rfam database比对注释,接着用cmsearch程序(参数默认)确定最终的sRNA。
snRNA:(small nuclearRNA,小核RNA),它是真核生物转录后加工过程中RNA剪接体(spilceosome)的主要成分。
miRNA:MicroRNA(miRNA)是在真核生物中发现的一类内源性的具有调控功能的非编码RNA,前体全长约90bp,其成熟miRNA大小长约20~25个核苷酸(nt)。miRNA广泛存在于真核生物中,是一组不编码蛋白质的短序列RNA,它本身不具有开放阅读框(ORF),对基因的表达具有调控作用。
sRNA、snRNA、miRNA的预测原理类似,首先用Rfam软件进行Rfam database比对注释,接着用其cmsearch程序(参数默认)确定最终的sRNA、snRNA、miRNA。
表6 ncRNA去冗余后的统计结果
| samples | 5S_rRNA number | 5S_rRNA average length | 5S_rRNA totle length | 5_8S_rRNA number | 5_8S_rRNA average length | 5_8S_rRNA totle length | 5_ureB_sRNA number | 5_ureB_sRNA average length | 5_ureB_sRNA totle length | Afu_182 number | Afu_182 average length | Afu_182 totle length | Afu_190 number | Afu_190 average length | Afu_190 totle length | Afu_198 number | Afu_198 average length | Afu_198 totle length | Afu_294 number | Afu_294 average length | Afu_294 totle length | Afu_298 number | Afu_298 average length | Afu_298 totle length | Afu_300 number | Afu_300 average length | Afu_300 totle length | Afu_304 number | Afu_304 average length | Afu_304 totle length | Afu_309 number | Afu_309 average length | Afu_309 totle length | Afu_335 number | Afu_335 average length | Afu_335 totle length | Afu_455 number | Afu_455 average length | Afu_455 totle length | Afu_513 number | Afu_513 average length | Afu_513 totle length | Afu_514 number | Afu_514 average length | Afu_514 totle length | Fungi_SRP number | Fungi_SRP average length | Fungi_SRP totle length | Fungi_U3 number | Fungi_U3 average length | Fungi_U3 totle length | Intron_gpI number | Intron_gpI average length | Intron_gpI totle length | LSU_rRNA_bacteria number | LSU_rRNA_bacteria average length | LSU_rRNA_bacteria totle length | LSU_rRNA_eukarya number | LSU_rRNA_eukarya average length | LSU_rRNA_eukarya totle length | RNase_MRP number | RNase_MRP average length | RNase_MRP totle length | SSU_rRNA_bacteria number | SSU_rRNA_bacteria average length | SSU_rRNA_bacteria totle length | SSU_rRNA_eukarya number | SSU_rRNA_eukarya average length | SSU_rRNA_eukarya totle length | TPP number | TPP average length | TPP totle length | Telomerase_Asco number | Telomerase_Asco average length | Telomerase_Asco totle length | U2 number | U2 average length | U2 totle length | U4 number | U4 average length | U4 totle length | U5 number | U5 average length | U5 totle length | U6 number | U6 average length | U6 totle length | snR191 number | snR191 average length | snR191 totle length | snR36 number | snR36 average length | snR36 totle length | snR44 number | snR44 average length | snR44 totle length | snR51 number | snR51 average length | snR51 totle length | snR73 number | snR73 average length | snR73 totle length | snR75 number | snR75 average length | snR75 totle length | snoR38 number | snoR38 average length | snoR38 totle length | snoZ13_snr52 number | snoZ13_snr52 average length | snoZ13_snr52 totle length | snosnR60_Z15 number | snosnR60_Z15 average length | snosnR60_Z15 totle length | snosnR61 number | snosnR61 average length | snosnR61 totle length | tRNA number | tRNA average length | tRNA totle length |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sallet_1 | 37 | 112.53 | 5213 | 12 | 152.21 | 1821 | 1 | 292.0 | 292 | 1 | 165.0 | 165 | 1 | 21.0 | 21 | 1 | 123.0 | 123 | 1 | 26.0 | 26 | 2 | 82.0 | 164 | 1 | 15.0 | 15 | 1 | 65.0 | 65 | 1 | 354.0 | 354 | 1 | 22.0 | 22 | 1 | 24.0 | 24 | 1 | 33.0 | 33 | 1 | 112.0 | 112 | 1 | 233.0 | 233 | 2 | 222.5 | 445 | 4 | 278.5 | 1114 | 1 | 3356.0 | 3356 | 15 | 2140.666667 | 32110 | 1 | 214.0 | 214 | 2 | 1632.5 | 3265 | 18 | 1255.611111 | 22601 | 4 | 145.25 | 581 | 1 | 21.0 | 21 | 4 | 86.25 | 345 | 1 | 135.0 | 135 | 1 | 213.0 | 213 | 4 | 55.25 | 221 | 1 | 221.0 | 221 | 1 | 211.0 | 211 | 1 | 123.0 | 123 | 1 | 111.0 | 111 | 1 | 21.0 | 21 | 1 | 84.0 | 84 | 2 | 81.0 | 162 | 1 | 123.0 | 123 | 1 | 106.0 | 106 | 1 | 12.0 | 12 | 120 | 101.95 | 12234 |
| sallet_5 | 22 | 111.33 | 5321 | 13 | 132.32 | 1213 | 1 | 221.0 | 221 | 1 | 121.0 | 121 | 1 | 34.0 | 34 | 1 | 121.0 | 121 | 1 | 63.0 | 63 | 2 | 21.0 | 42 | 1 | 67.0 | 67 | 1 | 63.0 | 63 | 1 | 662.0 | 662 | 1 | 35.0 | 35 | 1 | 44.0 | 44 | 1 | 45.0 | 45 | 1 | 214.0 | 214 | 1 | 321.0 | 321 | 2 | 221.0 | 442 | 4 | 261 | 1044 | 1 | 3321.0 | 3321 | 15 | 2147.333333 | 32210 | 1 | 437.0 | 437 | 2 | 1072.5 | 2145 | 16 | 1585.75 | 25372 | 3 | 78 | 234 | 1 | 33.0 | 33 | 2 | 222 | 444 | 1 | 625.0 | 625 | 1 | 332.0 | 332 | 2 | 122.5 | 245 | 1 | 225.0 | 225 | 1 | 332.0 | 332 | 1 | 138.0 | 138 | 1 | 101.0 | 101 | 1 | 44.0 | 44 | 1 | 44.0 | 44 | 2 | 34.0 | 68 | 1 | 96.0 | 96 | 1 | 122.0 | 122 | 1 | 23.0 | 23 | 117 | 86.31623932 | 10099 |
| sallet_6 | 57 | 113.24 | 5442 | 21 | 121.21 | 1653 | 1 | 244.0 | 244 | 1 | 113.0 | 113 | 1 | 55.0 | 55 | 1 | 222.0 | 222 | 1 | 62.0 | 62 | 2 | 33.0 | 66 | 1 | 86.0 | 86 | 1 | 72.0 | 72 | 1 | 222.0 | 222 | 1 | 55.0 | 55 | 1 | 43.0 | 43 | 1 | 43.0 | 43 | 1 | 221.0 | 221 | 1 | 221.0 | 221 | 2 | 124.5 | 249 | 4 | 223 | 892 | 1 | 3326.0 | 3326 | 15 | 2148 | 32220 | 1 | 342.0 | 342 | 2 | 1660.5 | 3321 | 12 | 1778.75 | 21345 | 2 | 110.5 | 221 | 1 | 35.0 | 35 | 4 | 168.75 | 675 | 1 | 213.0 | 213 | 1 | 213.0 | 213 | 3 | 180.6666667 | 542 | 1 | 321.0 | 321 | 1 | 211.0 | 211 | 1 | 221.0 | 221 | 1 | 212.0 | 212 | 1 | 33.0 | 33 | 1 | 53.0 | 53 | 2 | 47.0 | 94 | 1 | 22.0 | 22 | 1 | 234.0 | 234 | 1 | 33.0 | 33 | 125 | 88.896 | 11112 |
| sallet_4 | 72 | 114.42 | 5762 | 23 | 144.11 | 1887 | 1 | 276.0 | 276 | 1 | 177.0 | 177 | 1 | 75.0 | 75 | 1 | 165.0 | 165 | 1 | 12.0 | 12 | 2 | 56.0 | 112 | 1 | 66.0 | 66 | 1 | 46.0 | 46 | 1 | 241.0 | 241 | 1 | 67.0 | 67 | 1 | 31.0 | 31 | 1 | 32.0 | 32 | 1 | 105.0 | 105 | 1 | 225.0 | 225 | 2 | 245.0 | 445 | 4 | 228.5 | 914 | 1 | 3116.0 | 3116 | 15 | 2140 | 32100 | 1 | 356.0 | 356 | 2 | 1173 | 2346 | 16 | 2009.0625 | 32145 | 4 | 139 | 556 | 1 | 64.0 | 64 | 5 | 174.4 | 872 | 1 | 332.0 | 332 | 1 | 214.0 | 214 | 4 | 55.25 | 221 | 1 | 342.0 | 342 | 1 | 432.0 | 432 | 1 | 214.0 | 214 | 1 | 333.0 | 333 | 1 | 55.0 | 55 | 1 | 36.0 | 36 | 2 | 48.0 | 96 | 1 | 37.0 | 37 | 1 | 584.0 | 584 | 1 | 83.0 | 83 | 109 | 112.2110092 | 12231 |
| sallet_3 | 21 | 112.62 | 5213 | 21 | 154.45 | 1213 | 1 | 288.0 | 288 | 1 | 198.0 | 198 | 1 | 43.0 | 43 | 1 | 172.0 | 172 | 1 | 25.0 | 25 | 2 | 22.0 | 44 | 1 | 24.0 | 24 | 1 | 77.0 | 77 | 1 | 276.0 | 276 | 1 | 88.0 | 88 | 1 | 56.0 | 56 | 1 | 36.0 | 36 | 1 | 104.0 | 104 | 1 | 325.0 | 325 | 2 | 221.5 | 443 | 4 | 227 | 908 | 1 | 3446.0 | 3446 | 15 | 2614.666667 | 39220 | 1 | 358.0 | 358 | 2 | 2266 | 4532 | 14 | 1523.642857 | 21331 | 5 | 153 | 765 | 1 | 33.0 | 33 | 5 | 112.6 | 563 | 1 | 432.0 | 432 | 1 | 221.0 | 221 | 5 | 69 | 345 | 1 | 214.0 | 214 | 1 | 321.0 | 321 | 1 | 332.0 | 332 | 1 | 222.0 | 222 | 1 | 56.0 | 56 | 1 | 78.0 | 78 | 2 | 66.0 | 132 | 1 | 62.0 | 62 | 1 | 124.0 | 124 | 1 | 34.0 | 34 | 122 | 101.8934426 | 12431 |
| sallet_2 | 34 | 111.91 | 5887 | 16 | 151.41 | 1214 | 1 | 294.0 | 294 | 1 | 191.0 | 191 | 1 | 11.0 | 11 | 1 | 122.0 | 122 | 1 | 87.0 | 87 | 2 | 56.0 | 112 | 1 | 88.0 | 88 | 1 | 44.0 | 44 | 1 | 288.0 | 288 | 1 | 93.0 | 93 | 1 | 91.0 | 91 | 1 | 85.0 | 85 | 1 | 125.0 | 125 | 1 | 214.0 | 214 | 2 | 222.5 | 445 | 4 | 213.5 | 854 | 1 | 3356.0 | 3356 | 15 | 2646.666667 | 39700 | 1 | 437.0 | 437 | 2 | 1632.5 | 3265 | 12 | 2678.75 | 32145 | 6 | 147.8333333 | 887 | 1 | 27.0 | 27 | 4 | 140.5 | 562 | 1 | 221.0 | 221 | 1 | 332.0 | 332 | 2 | 160.5 | 321 | 1 | 321.0 | 321 | 1 | 342.0 | 342 | 1 | 213.0 | 213 | 1 | 101.0 | 101 | 1 | 87.0 | 87 | 1 | 84.0 | 84 | 2 | 63.0 | 126 | 1 | 88.0 | 88 | 1 | 321.0 | 321 | 1 | 55.0 | 55 | 111 | 120.1171171 | 13333 |
说明: samples:样本名称; * number:ncRNA数量; * average length:ncRNA平均长度; * totle length:ncRNA总长度。
3.4 基因功能分析
customer_files/04function_annotation/
├── 04.01common_database/
├────── 04.01.00all_result_count
│ ├── [物种].[样本].all_anno_count.svg [所有常见数据库注释结果汇总图]
│ └── [物种].[样本].all_anno_count.txt [各样本预测基因gff文件]
├────── 04.01.01GO_result
│ ├── [物种].[样本].dataPlot.go.alldata.txt [各样本GO数据库注释结果统计]
│ ├── [物种].[样本].dataPlot.go.txt [各样本GO数据库注释结果统计(前20个结果)]
│ └── [物种].[样本].GO_classes.svg [各样本GO数据库注释结果统计图]
├────── 04.01.02KEGG_result
│ ├── [物种].[样本].dataPlot.kegg.txt [各样本KEGG数据库注释结果统计]
│ └── [物种].[样本].Kegg_Classes.svg [各样本KEGG数据库注释结果统计图]
├────── 04.01.03eggNOG_result
│ ├── [物种].[样本].Eggnog.count.txt [各样本Eggnog数据库注释结果统计]
│ ├── [物种].[样本].Eggnog.result.emapper.annotations [各样本Eggnog数据库注释结果(该结果也包含GO和KEGG注释结果)]
│ └── [物种].[样本].Eggnog_plot.svg [各样本Eggnog数据库注释结果统计图]
├────── 04.01.04KOG_result
│ ├── [物种].[样本].diamond.KOG.result [各样本KOG数据库注释结果]
│ ├── [物种].[样本].KOG.fig.count.txt [各样本KOG数据库注释结果统计]
│ └── [物种].[样本].KOG_plot.svg [各样本KOG数据库注释结果统计图]
├────── 04.01.05NR_result
│ ├── [物种].[样本].nr_count.txt [各样本NR数据库注释结果统计]
│ ├── [物种].[样本].nr_plot.svg [各样本NR数据库注释结果统计图]
│ └── [物种].[样本].diamond.nr.result [各样本NR数据库注释结果]
├────── 04.01.06TCDB_result
│ ├── [物种].[样本].tcdb_plot.svg [各样本tcdb数据库注释结果统计图]
│ └── [物种].[样本].diamond.tcdb.result [各样本tcdb数据库注释结果]
├────── 04.01.07Pfam_result
│ └── [物种].[样本].Pfam.tblout.result [各样本Pfam数据库注释结果]
├────── 04.01.08Swiss_Prot_result
│ └── [物种].[样本].diamond.swissprot.result [各样本swissprot数据库注释结果]
├────── 04.01.09CAZy_result
│ ├── [物种].[样本].cazy_plot.svg [各样本cazy数据库注释结果图]
│ └── [物种].[样本].CAZyme.table [各样本cazy数据库注释结果]
├── 04.02Effector/
│ ├── [物种].[样本].antismash_result [各样本次级代谢基因簇分析结果]
│ ├── [物种].[样本].protein.oneline_summary.signalp5 [各样本分泌蛋白预测结果]
│ ├── [物种].[样本].diamond.p450.result [各样本P450数据库注释结果]
│ ├── [物种].[样本].tmhmm.txt [各样本分泌蛋白预测结果]
│ └── [物种].[样本].TNSS_count.txt [各样本分泌系统蛋白及T3SS效应蛋白预测结果]
└── 04.03Pathogenicity_analysis/
├── [物种].[样本].diamond.DFVF.result [各样本DFVF数据库注释结果]
├── [物种].[样本].diamond.PHI.result [各样本PHI数据库注释结果]
├── [物种].[样本].PHI_count.svg [各样本PHI数据库注释结果统计图]
└── [物种].[样本].PHI_count.txt [各样本PHI数据库注释结果统计]
目前提供注释的通用功能数据库主要有GO、KEGG、KOG、NR、Pfam和Swiss-Prot。
功能注释基本步骤如下:
1)将预测基因的蛋白序列与各功能数据库进行Diamond 比对(evalue ≤ 1e-5);
2)比对结果过滤:对于每一条序列的比对结果,选取 score 最高的比对结果进行注释。
本项目进行的编码基因的注释结果统计如下图所示:

图6 基因功能分析结果统计图
说明:
横坐标database为参与注释的各个数据库,纵坐标number of gene为各个数据库注释出来的基因数量。
3.4.1 常用数据库
3.4.1.1 GO数据库注释
GO的全称是Gene Ontology,是一套国际标准化的基因功能描述的分类系统。GO分为三大类:1)细胞组分(Cellular Component):用于描述亚细胞结构、位置和大分子复合物,如核仁、端粒和识别起始的复合物;2)分子功能(Molecular Function):用于描述基因、基因产物个体的功能,如与碳水化合物结合或 ATP 水解酶活性等;3)生物过程(Biological Process):用来描述基因编码的产物所参与的生物过程,如有丝分裂或嘌呤代谢等。
GO数据库三大分类统计结果如下图:

图7 GO数据库分类统计图
说明:
横坐标GO Term为注释到的GO Term,结果数量太多的只显示前20个。纵坐标Count为注释到各个GO Term的基因数量。图中不同颜色分别对应GO数据库中的BP、CC、MF三个分类。
3.4.1.2 KEGG数据库注释
KEGG全称为Kyoto Encyclopedia of Genes and Genomes。系统分析基因产物和化合物在细胞中的代谢途径以及这些基因产物的功能的数据库。它整合了基因组、化学分子和生化系统等方面的数据,包括代谢通路(KEGG PATHWAY)、药物(KEGG DRUG)、疾病(KEGG DISEASE)、功能模型(KEGG MODULE)、基因序列(KEGG GENES)及基因组(KEGG GENOME)等等。KO(KEGG ORTHOLOG)系统将各个KEGG注释系统联系在一起,KEGG已建立了一套完整KO注释的系统,可完成新测序物种的基因组或转录组的功能注释。详见http://www.genome.jp/kegg/。
绘制KEGG数据库中注释基因数目统计图如下:

图8 KEGG数据库分类统计图
说明:横坐标count(%)为注释到各个KEGG pathway的基因占所有基因中的百分比,纵坐标为注释到的KEGG pathway。
3.4.1.3 eggNOG数据库注释
eggNOG数据库,全称是evolutionary genealogy of genes: Non-supervised Orthologous Groups,是一个蛋白聚类数据库,带有功能描述和功能类别说明,由EMBL(欧洲分子生物实验室)维护。包含1,133个物种、721,801个直系同源组、41个不同水平的直系同源组分类,整合了5,214,234个蛋白序列。分别更新了4,873个COG数据库信息和4,850个KOG数据库信息。
eggNOG数据库按照功能一共可以分为二十五类,其统计结果如下图:

图9 eggNOG数据库分类统计图
说明:横坐标eggNOG classes为eggNOG数据库的不同分类,纵坐标count为注释到eggNOG数据库不同分类的基因数量。
3.4.1.4 KOG数据库注释
KOG数据库,属于COG数据库的一个针对真核生物的直系同源数据库。
COG,全称是Cluster of Orthologous Groups of proteins,由NCBI创建并维护的蛋白数据库,根据细菌、藻类和真核生物完整基因组的编码蛋白系统进化关系分类构建而成。通过比对可以将某个蛋白序列注释到某一个COG中,每一簇COG由直系同源序列构成,从而可以推测该序列的功能。COG数据库按照功能一共可以分为二十五类,详见http://www.ncbi.nlm.nih.gov/COG/。
COG数据库按照功能一共可以分为二十五类,其统计结果如下图:

图10 KOG数据库分类统计图
说明:横坐标KOG classes为KOG数据库不同分类,纵坐标count(%)为注释到不同KOG classes基因占所有基因的百分比。
3.4.1.5 NR数据库注释
NR全称为Non-Redundant Protein Database,是一个非冗余的蛋白质数据库,由NCBI创建并维护,其特点在于内容比较全面,同时注释结果中会包含有物种信息,可作物种分类用。根据基因注释到的物种情况,统计注释到的物种及基因数目,其统计结果如下图:

图11 NR数据库物种比对统计图
说明:横坐标species为基因在NR数据库注释到的不同物种,纵坐标count为注释到不同物种的基因数量。
3.4.1.6 TCDB数据库注释
TCDB,全称是Transporter Classification Database,转运蛋白分类数据库,是膜转运蛋白,包括离子通道(ion channels)的分类系统(TC system)。TCDB数据库转移系统以5个级别进行分类,第一级统计结果如下图:

图12 TCDB数据库功能分类统计图
说明:横坐标Function classes为TCDB数据库的不同功能分类,纵坐标Number of matched genes为注释到不同功能分类的基因数量。
3.4.1.7 Pfam数据库注释
蛋白质一般由一个或多个功能区构成,这些区通常被称为域。结构域的不同组合方式产生的蛋白质在自然界中各种不同。因此蛋白结构域的鉴别对分析蛋白质的功能来说尤其重要。Pfam数据库有两个组成部分:Pfam-A和Pfam-B,其中Pfam-A经过人工筛选,质量较高。详见http://pfam.xfam.org/。
该分析对预测基因对应的蛋白质序列进行Pfam注释比对,获得每个蛋白序列的结构域信息,结果在04.01common_database/文件夹下,文件名格式为:[物种].[样本].Pfam.tblout.result。
3.4.1.8 Swiss-Prot数据库注释
Swiss-Prot是一个精选的蛋白质序列数据库,它提供一个高水平的注释结果,例如一个蛋白质功能、其域结构、翻译后修饰、变异等的描述。详见http://www.ebi.ac.uk/uniprot/。
我们使用diamond软件对预测的蛋白质序列和Swiss-Prot数据库提供的蛋白质序列进行比对,从而对每个蛋白质序列进行注释,结果在04.01common_database/文件夹下,文件名格式为:[物种].[样本].diamond.swissprot.result。
3.4.1.9 碳水化合物活性酶(CAZy)数据库注释
CAZy全称为Carbohydrate-Active enZYmes Database,碳水化合物酶相关的专业数据库,内容包括能催化碳水化合物降解、修饰、以及生物合成的相关酶系家族。其包含五个主要分类:糖苷水解酶 (Glycoside Hydrolases, GHs)、糖基转移酶(GlycosylTransferases, GTs)、多糖裂解酶(Polysaccharide Lyases, PLs)和糖类酯解酶(Carbohydrate Esterases, CEs)、氧化还原酶(Auxiliary Activities, AAs)。
碳水化合物结合结构域是一种非催化结构域,能折叠成特定的三维空间结构,具有结合碳水化合物的功能。近年来研究表明:碳水化合物结合结构域能通过结合碳水化合物活性酶的底物,提高碳水化合物活性酶的催化结构域作用于底物的活性。
CAZy数据库分类注释结果个数统计图展示如下:

图13 CAZy数据库分类统计图
说明:横坐标Cazy Classes为Cazy数据库不同分类,纵坐标Number of matched genes为注释到各个分类的基因数量。
3.4.2 效应子
3.4.2.1 分泌蛋白预测
分泌蛋白是指在细胞内合成后,在信号肽的引导下穿过细胞膜分泌到细胞外起作用的蛋白质。分泌蛋白中有许多是生命活动所需的重要酶类。分泌蛋白的N端是由15~30个氨基酸组成的信号肽,对分泌蛋白的分泌起主导作用。
使用SignalP、TMHMM工具进行预测,检测是否含有信号肽及跨膜结构,综合预测蛋白序列是否是分泌蛋白。
表7 分泌蛋白预测结果
| sample ID | Signal Protein | TMHMM Protein | Secreted Protein |
|---|---|---|---|
| sallet_1 | 1349 | 2231 | 235 |
| sallet_2 | 1328 | 2447 | 123 |
| sallet_3 | 1276 | 2135 | 233 |
| sallet_4 | 1523 | 2251 | 256 |
| sallet_5 | 1763 | 2664 | 276 |
| sallet_6 | 1523 | 2213 | 289 |
说明: sample ID:样本名称; Signal Protein:Signal软件预测的含有信号肽的蛋白质数量; TMHMM Protein:TMHMM软件预测的含有跨膜结构的蛋白质数量; Secreted Protein:综合预测为分泌蛋白的数量。
3.4.2.2 分泌系统蛋白及T3SS效应蛋白预测
病原菌通过分泌系统TNSS(type N secretion systems,目前确定的有7种,I型-VII型)将该类蛋白分泌至胞外或是宿主细胞中,通过控制免疫应答反应以及细胞衰亡引起病理反应,而其中革兰氏阴性菌的T3SS通常用来从分子水平研究病原菌,感染机制,毒力作用等,是研究得比较多的分泌系统。
对于TNSS系统,通过蛋白序列功能数据库注释结果中,提取分泌系统相关蛋白进行注释。对于革兰氏阴性菌,另外采用EffectiveT3 软件预测T3SS效应蛋白。
表8 TNSS结果统计
| sample ID | totle gene | T1SS num | T2SS num | T3SS num | T4SS num | T5SS num | T6SS num | T7SS num |
|---|---|---|---|---|---|---|---|---|
| sallet_1 | 119330 | 1 | 12 | 12 | 23 | 12 | 26 | 27 |
| sallet_2 | 128832 | 2 | 23 | 22 | 21 | 22 | 27 | 73 |
| sallet_3 | 119221 | 2 | 21 | 23 | 24 | 26 | 34 | 32 |
| sallet_4 | 132123 | 12 | 32 | 34 | 27 | 21 | 31 | 87 |
| sallet_5 | 118872 | 23 | 22 | 21 | 26 | 24 | 26 | 73 |
| sallet_6 | 121345 | 21 | 21 | 24 | 27 | 27 | 28 | 26 |
说明: sample ID:样本名称; totle gene:所有基因数量; T1SS num:T1SS数量; T2SS num:T2SS数量; T3SS num:T3SS数量; T4SS num:T4SS数量; T5SS num:T5SS数量; T6SS num:T6SS数量; T7SS num:T7SS数量;
表9 T3SS效应蛋白预测结果统计
| sample ID | Totle gene | T3SS effective true | T3SS effective false |
|---|---|---|---|
| sallet_1 | 23866 | 2633 | 21233 |
| sallet_2 | 12563 | 6266 | 6297 |
| sallet_3 | 16553 | 7373 | 9180 |
| sallet_4 | 17873 | 2183 | 15690 |
| sallet_5 | 12222 | 3741 | 8481 |
| sallet_6 | 23631 | 283 | 23348 |
说明: sample ID:样本名称; Totle gene:所有基因数量; T3SS effective true:预测为T3SS效应蛋白的数量; T3SS effective false:预测不是T3SS效应蛋白的数量;
3.4.2.4 次级代谢基因簇分析
次级代谢产物是微生物在一定的生长时期,以初级代谢产物为前体合成的对微生物的生命活动无明确功能,并非生长繁殖所必需的物质。采用antiSMASH程序对基因组进行预测。
PKS可分为三种类型:I型也成为模块类PKS,是由s多个结构域组成的多功能酶复合物。II型也成为芳香类PKS,主要合成芳香类化合物。III型也成查尔酮型PKS。使用antiSMASH程序对基因组进行预测。
表10 次级代谢基因簇及基因数量统计
| sample ID | NRPS_cluster_number | NRPS_gene_number | NRPS-like_cluster_number | NRPS-like_gene_number | T1PKS_cluster_number | T1PKS_gene_number | T3PKS_cluster_number | T3PKS_gene_number | fungal-RiPP_cluster_number | fungal-RiPP_gene_number | terpene_cluster_number | terpene_gene_number |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sallet_1 | 4 | 21 | 3 | 23 | 21 | 120 | 21 | 123 | 3 | 6 | 12 | 27 |
| sallet_2 | 6 | 23 | 4 | 34 | 22 | 231 | 21 | 123 | 6 | 18 | 18 | 37 |
| sallet_3 | 4 | 34 | 8 | 123 | 22 | 221 | 12 | 66 | 8 | 16 | 28 | 48 |
| sallet_4 | 3 | 57 | 12 | 156 | 11 | 221 | 15 | 98 | 10 | 29 | 38 | 55 |
| sallet_5 | 5 | 36 | 7 | 120 | 16 | 216 | 17 | 37 | 11 | 33 | 21 | 54 |
| sallet_6 | 7 | 57 | 6 | 55 | 16 | 123 | 12 | 28 | 19 | 48 | 22 | 38 |
说明: sample ID:样本名称; *_cluster_number:某类次级代谢产物聚类数量; *_gene_number:某类次级代谢产物基因数量。

图14 各类PKS基因簇及基因数量统计
说明:横坐标PKS type为不同PKS类型,纵坐标count为注释到不同PKS类型的基因数量。
3.4.2.5 P450数据库注释
细胞色素P450(cytochromeP450或CYP450,简称CYP450)为一类亚铁血红素—硫醇盐蛋白的超家族,它参与内源性物质和包括药物、环境化合物在内的外源性物质的代谢。
我们使用diamond软件对预测的蛋白序列进行P450数据库注释,获得每个蛋白序列对应的P450信息,结果在04.02Effector/文件夹下,文件名格式为:[物种].[样本].diamond.p450.result。
3.4.3 致病性分析
3.4.3.1 病原与宿主互作数据库(PHI)注释
PHI全称为Pathogen Host Interactions Database,病原与宿主互作数据库,主要来源于真菌、卵菌和细菌病原,感染的宿主包括动物、植物、真菌以及昆虫。该数据库对寻找药物干预的靶基因研究有重要作用,同时该数据库还包括抗真菌化合物和相应的靶基因。数据库中的每个基因都包含核酸和氨基酸序列,以及感染宿主过程中预测的蛋白功能的详细描述。
病原体PHI表型突变类型基因数目的统计情况如下图所示:

图15 病原体PHI表型突变类型统计
说明:横坐标mutation type为不同的病原PHI表型突变类型,纵坐标number of genes为注释到不同突变类型的基因数量。
3.4.3.2 真菌毒力因子数据库(DFVF)注释
DFVF数据库全称为database of fungal virulence factors(真菌毒力因子数据库),是一个综合的已知真菌毒力因子数据库,收集了来自85个属的228个真菌菌株所产生的2058个致病基因。每个基因详细描述引起的疾病和作用的宿主,更与Pfam功能域注释和GO注释信息进行了关联。
我们使用Diamond软件,把目标物种的氨基酸序列,与DFVF数据库进行比对,把目标物种的基因和其相对应的功能注释信息结合起来,得到注释结果,结果在04.03Pathogenicity_analysis/文件夹下,文件名格式为:[物种].[样本].diamond.DFVF.result。
3.5 比较基因组分析
customer_files/05Comparative_Genomics_Analysis
├── 05.01Collinearity_analysis/
│ ├── [物种].[样本].plot.svg [各样本基因组共线性图]
│ ├── [物种].[样本].plotsr.pdf [各样本基因组共线性图]
│ ├── [物种].[样本].syri.out [各样本基因组结构差异文件]
│ └── [物种].[样本].syri.vcf [各样本基因组结构差异文件]
├── 05.02SNP/
│ └── [样本].snp [各样本SNP识别结果]
├── 05.03Indel/
│ └── [物种].[样本]_indel.result [各样本Indel识别结果]
├── 05.04SV/
│ └── [样本].raw.vcf [各样本SV识别结果]
└── 05.04SV/
└── [样本].sv.svg [各样本SV识别结果可视化]
3.5.1 全基因组共线性分析
共线性指的是遗传学中的基因连锁关系, 是不同物种基因组上同源基因以相同顺序排列的现象。两个物种之间的共线性程度可以作为衡量他们之间进化距离的尺度,可以知道物种间的亲缘关系。
使用MUMmer软件对样本基因组和参考基因组进行比对,确定样本基因组和参考基因组之间的比对关系。然后使用SyRI软件识别样本基因组和参考基因组之间的共线性区域。
样品基因组和参考基因组之间的共线性展示结果如下图:

图16 基因组共线性线图
说明:横坐标和纵坐标分别表示比对的两个基因组位置,图中的连线为两个基因组共线性部分。

图17 基因组共线性比对图
说明:横坐标Chromosome postion(in Mbp)表示基因组上的位置,单位为Mb。纵坐标Reference Chromosome ID为参考基因组的不同contig,每个contig ID对应的上下两条横线分别表示参考基因组和对比基因组的contig。
3.5.2 SNP统计与注释
SNP(单核苷酸多态性)主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性,包括单个碱基的转换、颠换等。
采用MUMmer比对软件进行个体SNP的检测,依照SNP和基因之间的位置关系及相互作用,对SNP功能进行注释。
表11 SNP结果统计
| Reference Name | sample ID | Start_syn | Stop_syn | Start_nonsyn | Stop_nonsyn | Premature_stop | Synonymous | Nonsynonymous | Total_CDS_SNP | Intergenic | Total_SNP | GCF_000146045 | sallet_1 | 12 | 21 | 23 | 11 | 22 | 12 | 34 | 44 | 21 | 21 | GCF_000146045 | sallet_2 | 20 | 32 | 21 | 12 | 44 | 12 | 2 | 45 | 21 | 119 | GCF_000146045 | sallet_3 | 23 | 1 | 2 | 21 | 2 | 2 | 2 | 32 | 23 | 110 | GCF_000146045 | sallet_4 | 12 | 1 | 2 | 23 | 3 | 1 | 3 | 22 | 24 | 92 | GCF_000146045 | sallet_5 | 21 | 2 | 1 | 23 | 3 | 2 | 2 | 33 | 32 | 119 | GCF_000146045 | sallet_6 | 12 | 1 | 2 | 23 | 3 | 2 | 2 | 44 | 22 | 111 |
|---|
说明: Reference Name:参考序列ID; Sample_name:样品ID; Start_syn:起始密码子同义突变; Stop_syn:终止密码子同义突变; Start_nonsyn:起始密码子非同义突变; Stop_nonsyn:终止密码子非同义突变; Premature_stop:无义突变,该位点三联体密码子突变成终止密码子; Synonymous:基因区内同义突变; Nonsynonymous:基因区内的非同义突变; Total_CDS_SNP:位于基因区的SNP; Intergenic:位于基因间区的SNP; Total_SNP:样品总SNP。
3.5.3 InDel统计与注释
InDel是指基因组中小片段的插入和缺失序列。
利用LASTZ软件检测长度小于50bp的小片段插入与缺失(InDel),对InDel类型进行统计,结果见下表:
表12 InDel类型统计
| Reference Name | sample ID | Start codon ins | CDS inside ins | Stop codon ins | Start codon del | CDS inside del | Stop codon del | GCF_000146045 | sallet_1 | 12 | 1 | 3 | 23 | 3 | 2 | GCF_000146045 | sallet_2 | 20 | 1 | 2 | 23 | 3 | 2 | GCF_000146045 | sallet_3 | 23 | 1 | 2 | 21 | 2 | 2 | GCF_000146045 | sallet_4 | 12 | 1 | 2 | 23 | 3 | 1 | GCF_000146045 | sallet_5 | 21 | 2 | 1 | 23 | 3 | 2 | GCF_000146045 | sallet_6 | 12 | 1 | 2 | 23 | 3 | 2 |
|---|
说明 Reference Name:参考序列ID; Sample ID:样品ID; Start codon ins:位于起始密码子的插入; CDS inside ins:位于CDS中间的插入; Stop codon ins:位于终止密码子的插入; Start codon del:位于起始密码子的缺失; CDS inside del:位于CDS中间的缺失; Stop codon del:位于终止密码子的缺失。
对InDel进行注释,注释结果见下表:
表13 InDel注释结果统计
| Reference Name | sample ID | Frame-shifted | Start codon | Stop codon | Premature stop | Effect | CDS with indel | All CDS | GCF_000146045 | sallet_1 | 12 | 1 | 3 | 23 | 3 | 2 | 200 | GCF_000146045 | sallet_2 | 20 | 1 | 2 | 23 | 3 | 2 | 200 | GCF_000146045 | sallet_3 | 23 | 1 | 2 | 21 | 2 | 2 | 200 | GCF_000146045 | sallet_4 | 12 | 1 | 2 | 23 | 3 | 1 | 200 | GCF_000146045 | sallet_5 | 21 | 2 | 1 | 23 | 3 | 2 | 200 | GCF_000146045 | sallet_6 | 12 | 1 | 2 | 23 | 3 | 2 | 200 |
|---|
说明 Reference Name:参考序列ID; Sample ID:样品ID; Frame-shifted:ORF移码突变; Start codon:ORF起始密码子被破坏; Stop codon:ORF终止密码子被破坏; Premature stop:提前终止了ORF; Effect:对ORF无重大影响; CDS with indel:存在InDel的CDS个数; All CDS:参考序列的CDS总个数。
3.5.4 SV统计与注释
基因组结构变异(Structural Variation,SV)一般是指基因组上大长度的序列变化和位置关系变化。基因组结构变异包含很多种类型,通常定义是长度大于50bp的插入(Insertion)、缺失(Deletion)、串联重复(Tandem repeate)、染色体倒位(Inversion)、染色体内部或染色体之间的序列易位(Translocation)、拷贝数变异(CNV)以及形式更为复杂的嵌合性变异。其中,占比最大的就是大片段的插入删除。
使用MUMmer软件对样本基因组和参考基因组进行比对,确定样本基因组和参考基因组之间的比对关系。然后使用SyRI软件识别样本基因组中的结构变异信息。
全基因组结构变异类型配对图如下所示:

图18 结构变异可视化
说明 红色(圆环):样本基因组; 绿色(圆环):参考基因组; 橙色:大片段插入; 蓝色:易位; 绿色:大片段删除; 粉色:倒置;
3.6 群体进化分析
customer_files/06population_evolution/ ├── 06.01cor_pan_genes/ │ ├── [物种].cd_hit.result.clstr [各样本蛋白质聚类结果] │ └── Flower_plot.svg [各样本共有基因及特意基因花瓣图] └── 06.02Hcluster/ ├── gene_family_bar_count.txt [各样本基因家族柱状图数据] ├── gene_family_count.txt [各样本基因家族统计] ├── orthologs_gene_bar_plot.svg [同源基因柱状图] ├── Phylogenetic_Tree.svg [系统发生树] └── Phylogenetic_Tree_dis.svg [系统发生树带距离]
3.6.1 共有和特有基因分析
所有样本中均存在的同源基因称为共有基因(Core gene),除去共有基因,其他的基因称为非共有基因(Dispensable gene),特有基因(Specific gene)是只有在某个样品中所特异拥有的基因。其中共有基因(Core gene)和特有基因(Specific gene)很可能与样品的共性和特性相对应,可以作为样本间功能差异的研究依据。
使用cd-hit软件对需要分析的多个样品的蛋白序列进行聚类,并用R进行绘图。
通过比较多个样品的蛋白质序列,我们统计了它们的共有基因和特有基因,以花瓣图形式展示:
图19 共有基因和特有基因花瓣图
说明:花瓣图中不同花瓣表示各个样本中特异的基因数量,花瓣中心为所有样本共有基因数量。
3.6.2 基因家族分析
基因组进化中,一个基因通过基因重复产生了两个或更多的拷贝,这些基因即构成一个基因家族。基因家族是具有共同祖先的一组基因,家族内不同基因往往具有相似的结构和功能。
使用diamond软件对多个目标基因组的蛋白进行两两比对,过滤比对不可信的结果,再使用Solar(Version 0.9.6)去除冗余,用Hcluster-sg按照比对相似度对蛋白进行聚类,得到基因家族聚类结果。
表14 基因家族鉴定的统计结果
| samples | totle gene number | gene number in families | uncluster genes number | family number | unique family number |
|---|---|---|---|---|---|
| sallet_1 | 11933 | 11926 | 7 | 11241 | 10798 |
| sallet_2 | 11933 | 11923 | 10 | 11238 | 10794 |
| sallet_3 | 11933 | 11924 | 9 | 11239 | 10797 |
| sallet_4 | 11933 | 11925 | 8 | 11241 | 10798 |
| sallet_5 | 11933 | 11926 | 7 | 11241 | 10799 |
| sallet_6 | 11933 | 11923 | 51 | 11240 | 10797 |
说明: samples:样本名称; totle gene number:所有基因数量; gene number in families:可归到基因家族中基因数量; uncluster genes number:未归到聚类中基因数量; family number:样本中基因家族数量; unique family number:唯一家族数量
图20 同源基因数目统计条形图
说明:横坐标samples为各个样本名称,纵坐标Number of genes为各个样本不同基因家族中的基因数量。
图21 多个物种/样本的同源基因家族数目Venn图
3.6.3 物种进化分析
系统发生树(英文:phylogenetic tree或evolutionary tree)是表明被认为具有共同祖先的各物种相互间演化关系的树。 它用来表示系统发生研究的结果,用它描述物种之间的进化关系。
用Treebest(Version 1.9.2)(Neighbor-Joining,NJ)或 PHYML(Maximum likelihood,ML)(Version v3.0)软件构建进化树,物种之间的进化树见下图:
图22 物种进化系统发生树
说明:该图为系统发生树,即进化树,图中各个样本名字所在的位置为进化树的叶子节点。图中分叉位置标注的数字为进化树的支持度,该数值在0%-100%之间,值越大说明越多证据支持该分支。
3.7 甲基化分析
customer_files/07methylation/ ├── 07.01modification/ │ ├── [样本].basemods.gff [各样本识别的甲基化结果] │ ├── [物种].modifications_count.txt [所有样本识别的甲基化分类统计] │ ├── [样本].motif.gff [各样本识别的甲基化motif结果] │ └── [物种].motif_count.txt [所有样本识别的甲基化motif结果统计] ├── 07.02motif_GR_IGR/ │ └── [物种].motif_GR_IGR_count.txt [所有样本识别的甲基化motif在GR/IGR区域统计] ├── 07.03unmodification_motif_GR_IGR/ │ └── [物种].unmodification_motif_GR_IGR_count.txt [所有样本识别的未甲基化motif在GR/IGR区域统计] ├── 07.04motif_gene_anno/ │ ├── [物种].motif_gene_anno.txt [所有样本motif基因注释结果统计] │ ├── [物种].[样本].augustus.protein.oneline_summary.signalp5 [各样本motif基因secretory注释结果] │ ├── [物种].[样本].diamond.DFVF.result [各样本motif基因DFVF注释结果] │ ├── [物种].[样本].diamond.KOG.result [各样本motif基因KOG注释结果] │ ├── [物种].[样本].diamond.nr.result [各样本motif基因NR注释结果] │ ├── [物种].[样本].diamond.PHI.result [各样本motif基因PHI注释结果] │ ├── [物种].[样本].diamond.swissprot.result [各样本motif基因swissprot注释结果] │ ├── [物种].[样本].diamond.tcdb.result [各样本motif基因tcdb注释结果] │ └── [物种].[样本].Eggnog.result.emapper.annotations [各样本motif基因GO和KEGG注释结果] ├── 07.05COG_anno/ │ ├── [样本].COG.fig.count.txt [各样本motif序列COG注释结果统计] │ └── [样本].COG_plot.svg [各样本motif序列COG注释结果统计图] └── 07.06circos/ └── [样本].circos.svg [各样本甲基化circos图]
3.7.1 表观修饰
使用SMRT Link,对最终的基因组组装结果进行甲基化位点检测和可能的甲基化转移酶识别的核苷酸基序(motif)的预测。能够预测到的修饰类型包括m6A,m4C和m5C,以及未知类型(modified_base)。
表15 样品基因组修饰位点各类型统计信息
| samples | m4C number | m4C percent(%) | m6A number | m6A percent(%) | modified_base number | modified_base percent(%) |
|---|---|---|---|---|---|---|
| sallet_1 | 27382 | 4.208840379 | 1123 | 0.172614409 | 622078 | 95.61854521 |
| sallet_2 | 27162 | 3.551001358 | 1287 | 0.168254869 | 736462 | 96.28074377 |
| sallet_3 | 26631 | 4.016093934 | 1264 | 0.190617804 | 635212 | 95.79328826 |
| sallet_4 | 17263 | 3.109408378 | 1652 | 0.297557936 | 536271 | 96.59303369 |
| sallet_5 | 26317 | 3.966187113 | 1736 | 0.261629397 | 635481 | 95.77218349 |
| sallet_6 | 28374 | 3.703839075 | 1284 | 0.167608704 | 736412 | 96.12855222 |
说明: samples:样本名称; m4C number:m4C碱基数量; m4C percent(%):m4C碱基占所有碱基百分比; m6A number:m6A碱基数量; m6A percent(%):m6A碱基占所有碱基百分比; modified_base number:所有甲基化修饰碱基数量; modified_base percent(%):所有甲基化修饰碱基数量占所有碱基百分比。
表16 样品基因组基序motif统计信息
| sampleID | motifString | centerPos | ModificationType | fraction | nDetected | meanScore | objectiveScore |
|---|---|---|---|---|---|---|---|
| sallet_1 | CAGNNNNNNNNTCTY | 1167 | 6mA | 38 | 1156 | 32 | 56 |
| sallet_2 | AGANNNNNNNNCTG | 1167 | 6mA | 33 | 1156 | 35 | 90 |
| sallet_3 | GAACAC | 1167 | 6mA | 20 | 1056 | 36 | 98 |
| sallet_4 | GCGGCCGC | 1167 | 6mA | 21 | 156 | 45 | 100 |
| sallet_5 | GCNNNNNNNGC | 1167 | 6mA | 38 | 1156 | 34 | 76 |
| sallet_6 | GCNNNNNNNGCGGGG | 14447 | 4mA | 21 | 1226 | 34 | 82 |
说明 motifString: 甲基转移酶识别的核苷酸motif序列; centerPos:修饰碱基所在的位置; ModificationType:修饰类型; fraction:修饰的motif占基因组中全部此motif的比例; nDetected:修饰的motif个数; meanScore:平均得分; objectiveScore:全部motif的总得分值。
3.7.2 甲基化motif在GR/IGR上的分布统计
表17 甲基化motif在GR/IGR上的分布统计信息
| sampleID | m4C.ingenome | m4C.inGR(%) | m4C.inIGR(%) | 5mC.ingenome | m5C.inGR(%) | m5C.inIGR(%) | m6A.ingenome | m6A.inGR(%) | m6A.inIGR(%) | modified_base.ingenome | modified_base.inGR(%) | modified_base.inIGR(%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sallet_1 | 2211 | 20.62 | 79.38 | 1111 | 39.96 | 60.04 | 1234 | 27.07 | 72.93 | 4556 | 27.09 | 72.91 |
| sallet_2 | 2132 | 21.39 | 78.61 | 2342 | 18.96 | 81.04 | 2221 | 15.04 | 84.96 | 6695 | 18.43 | 81.57 |
| sallet_3 | 1234 | 36.95 | 63.05 | 3214 | 13.81 | 86.19 | 2555 | 13.07 | 86.93 | 7003 | 17.62 | 82.38 |
| sallet_4 | 2222 | 20.52 | 79.48 | 3421 | 12.98 | 87.02 | 2251 | 14.84 | 85.16 | 7894 | 15.63 | 84.37 |
| sallet_5 | 1232 | 37.01 | 62.99 | 2134 | 20.81 | 79.19 | 2213 | 15.09 | 84.91 | 5579 | 22.12 | 77.88 |
| sallet_6 | 1232 | 37.01 | 62.99 | 2222 | 19.98 | 80.02 | 3214 | 10.39 | 89.61 | 6668 | 18.51 | 81.49 |
说明 m4C. in genome:m4C甲基化类型在基因组上的数量; m4C. in GR(%):m4C甲基化类型在基因区域上的数量; m4C. in IGR(%):m4C甲基化类型在基因间区上的数量; 5mC. in genome:m5C甲基化类型在基因组上的数量; m5C. in GR(%):m5C甲基化类型在基因区域上的数量; m5C. in IGR(%):m5C甲基化类型在基因间区上的数量; m6A. in genome:m6A甲基化类型在基因组上的数量; m6A. in GR(%):m6A甲基化类型在基因区域上的数量; m6A. in IGR(%):m6A甲基化类型在基因间区上的数量; modified_base. in genome: 未知类型甲基化类型在基因组上的数量; modified_base. in GR(%):未知类型甲基化类型在基因区域上的数量; modified_base. in IGR(%):未知类型甲基化类型在基因间区上的数量。
3.7.3 未甲基化motif在GR/IGR上的分布统计
表18 未甲基化motif在GR/IGR上的分布统计信息
| sampleID | motifString | No.ingenome | No.inGR(%) | No.inIGR(%) |
|---|---|---|---|---|
| sallet_1 | GCNNNNNNNGCGGGG | 1111 | 20.62 | 79.38 |
| sallet_2 | GCNNGCGCGCGGGG | 1221 | 20.32 | 79.68 |
| sallet_3 | GCCCCNNNNGCGGGG | 2221 | 25.62 | 74.38 |
| sallet_4 | GCGCGCNNNNGCGGGG | 4426 | 10.42 | 89.58 |
| sallet_5 | GCCCNNGCGGGG | 1234 | 20.62 | 79.38 |
| sallet_6 | GCCNNNGCGGGG | 3342 | 27.62 | 72.38 |
说明 motifString:未甲基化的甲基转移酶识别的核苷酸motif序列; No. in genome:未甲基化motif序列在基因组上的数量; No. in GR(%):未甲基化motif序列在基因区域上的数量; No. in IGR(%):未甲基化motif序列在基因间区上的数量。
3.7.4 motif基因注释
样品各motif位点上的基因在NR,SwissProt,KEGG,GO等各大数据库的注释情况展示。
表19 motif基因注释
| sampleID | motifString | nr | swissprot | KOG | tcdb | PHI | DFVF | cazy | secretory | GO | KEGG |
|---|---|---|---|---|---|---|---|---|---|---|---|
| sallet_1 | GCNNNNNNNGCGGGG | 1112 | 123 | 1124 | 234 | 345 | 451 | 236 | 234 | 443 | 231 |
| sallet_2 | GCNNGCGCGCGGGG | 2134 | 2123 | 2213 | 2222 | 234 | 324 | 443 | 231 | 234 | 333 |
| sallet_3 | GCCCCNNNNGCGGGG | 2134 | 2123 | 2213 | 2222 | 234 | 324 | 443 | 231 | 234 | 333 |
| sallet_4 | GCGCGCNNNNGCGGGG | 2134 | 2123 | 2213 | 2222 | 234 | 324 | 443 | 231 | 234 | 333 |
| sallet_5 | GCCCNNGCGGGG | 2134 | 2123 | 2213 | 2222 | 234 | 324 | 443 | 231 | 234 | 333 |
| sallet_6 | GCCNNNGCGGGG | 2134 | 2123 | 2213 | 2222 | 234 | 324 | 443 | 231 | 234 | 333 |
说明: sampleID:样本名称; motifString:motif序列; nr:motif序列在nr数据库注释到的结果; swissprot:motif序列在swissprot数据库注释到的结果; KOG:motif序列在KOG数据库注释到的结果; tcdb:motif序列在tcdb数据库注释到的结果; PHI:motif序列在PHI数据库注释到的结果; DFVF:motif序列在DFVF数据库注释到的结果; cazy:motif序列在cazy数据库注释到的结果; secretory:motif序列在secretory数据库注释到的结果; GO:motif序列在GO数据库注释到的结果; KEGG:motif序列在KEGG数据库注释到的结果。
3.7.5 COG注释分布图
甲基转移酶识别的核苷酸motif序列的COG注释和绘图分析。

图23 甲基化motif序列COG注释结果
说明:横坐标KOG classes为COG数据库中不同的功能分类,纵坐标count(%)为注释到对应功能分类中的基因占所有基因的百分比。
3.7.6 甲基化圈图
表观修饰分布圈图如下所示:

图24 甲基化圈图
说明:
图中各圈从外至内依次代表:基因组位置,正义链修饰位点分布情况,m4C修饰位点数量,m5C修饰位点数量,m6C修饰位点数量,GC含量。
四、所用软件的版本
| 软件 | 版本 |
|---|---|
| Trimmomatic | 0.39 |
| FastQC | 0.11.9 |
| jellyfish | 2.2.10 |
| bedtools | 2.30.0 |
| canu | 2.2 |
| samtools | 1.7 |
| pbmm2 | 1.9.0 |
| minimap2 | 2.15 |
| RepeatModeler | 2.0.3 |
| tRNAscan-SE | 2.0.9 |
| diamond | 0.8.22 |
| SignalP | 5.0b |
| TMHMM | 2.0 |
| antiSMASH | 6.1.1 |
| MUMmer4 | 4.0.0 |
| lastz | 1.04.15 |
| OrthoFinder | 2.5.4 |
| ipdSummary | 3.0 |
五、 参考文献
1. Trimmomatic: a flexible trimmer for Illumina sequence data. (Trimmomatic)
2. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. (jellyfish)
3. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. (canu)
4. The Sequence Alignment/Map format and SAMtools. (samtools)
5. Minimap2: pairwise alignment for nucleotide sequences. (Minimap2)
6. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. (tRNAscan-SE)
7. Sensitive protein alignments at tree-of-life scale using DIAMOND. (diamond)
8. SignalP 5.0 improves signal peptide predictions using deep neural networks. (SignalP)
9. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. (TMHMM)
10. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. (antiSMASH)
11. MUMmer4: A fast and versatile genome alignment system. PLoS computational biology. (MUMmer4)
12. Improved pairwise alignment of genomic DNA. (LASTZ)
13. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. (OrthoFinder)