温馨提示:请使用火狐或者Chrome的网页浏览器来查看报告
微科盟真核有参转录组数据分析结题报告
一 概述
转录组是指特定组织或细胞在某个时间或某个状态下转录出来的所有RNA的总和,主要包括mRNA和非编码RNA。转录组测序是基于Illumina测序平台,研究特定组织或细胞在某个时期转录出来的所有mRNA,是基因功能与结构研究的基础,对理解生物体的发育和疾病的发生具有重要作用。随着基因测序技术的发展与测序成本的降低,RNA-Seq凭借高通量、高灵敏度、应用范围广等优势,已经成为转录组研究的主要方法。
真核有参转录组测序采用Illumina测序平台,对真核生物的所有RNA进行测序,将高质量数据与参考基因组进行比对,进一步进行表达定量、功能注释、变异分析等分析。转录组研究是基因功能及结构研究的基础和出发点,真核有参转录组物种的转录水平变化、分子机制及调控网络研究提供了有力的技术手段,目前已广泛应用于基础研究、临床诊断、药物研发和分子育种等领域。
二 项目流程
2.1 测序实验流程
从RNA样品提取到最终数据获得,样品检测、建库、测序等每一环节都会直接影响数据的数量和质量,从而影响后续信息分析的结果。为从源头保证测序数据准确可靠,我们承诺在数据的所有生产环节都严格把关,从根源上确保高质量数据的产出。建库测序的流程图如下(图2.1):
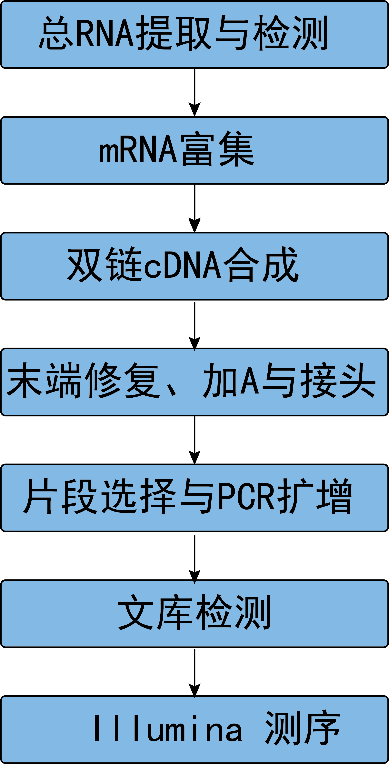
图2.1 RNA-Seq建库测序流程
2.1.1 RNA提取与检测
采用标准提取方法从组织或细胞中提取RNA,随后对RNA样品进行严格质控,质控使用Agilent 2100 bioanalyzer:精确检测RNA完整性。
2.1.2 文库构建与质检
文库构建
mRNA的获取主要有两种方式:一是利用真核生物大部分mRNA都带有多聚腺苷酸尾的结构特征,通过连接有寡聚胸腺嘧啶磁珠富集带有多聚腺苷酸尾的mRNA。二是从总RNA中去除核糖体RNA,从而得到剩余的RNA。随后在NEB Fragmentation Buffer中用二价阳离子将得到的mRNA随机打断,按照NEB普通建库方式或链特异性建库方式进行建库(图2.2)。
- NEB普通建库:以片段化的mRNA为模版,随机寡核苷酸为引物,在M-MuLV逆转录酶体系中合成cDNA第一条链,随后用RNaseH(RNA酶H)降解RNA链,并在DNA polymerase I (DNA聚合酶I)体系下,以dNTPs(四种脱氧核糖核苷酸)为原料合成cDNA第二条链。纯化后的双链cDNA经过末端修复、加腺苷酸尾并连接测序接头,用AMPure XP 磁珠筛选250-300bp左右的cDNA,进行PCR扩增并再次使用AMPure XP 磁珠纯化PCR产物,最终获得文库。建库原理如下面左图所示。
- 链特异性建库:逆转录合成cDNA第一条链方法与NEB普通建库方法相同,不同之处在于合成第二条链时,dNTPs中的dTTP由dUTP取代,之后同样进行cDNA末端修复、加A尾、连接测序接头和长度筛选,然后先使用USER酶降解含U的cDNA第二链再进行PCR扩增并获得文库。链特异性文库具有诸多优势,如相同数据量下可获取更多有效信息;能获得更精准的基因定量、定位与注释信息;能提供反义转录本及每一isoform中单一exon的表达水平。建库原理如下面右图所示:
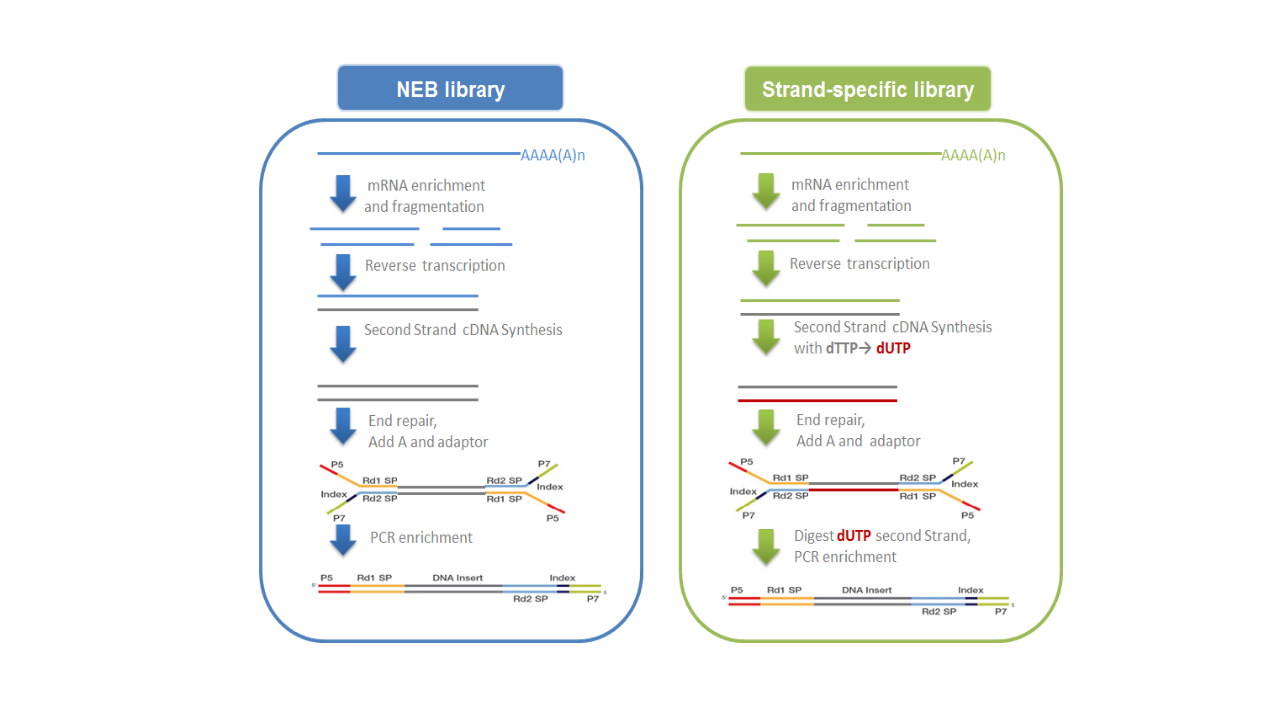
图2.2 文库构建原理示意图
测序接头:包括P5/P7,index和Rd1/Rd2 SP三个部分(如上图所示)。其中P5/P7是PCR扩增引物及flow cell上引物结合的部分,index提供区分不同文库的信息,Rd1/Rd2 SP即read1/read2 sequence primer,是测序引物结合区域,测序过程理论上由Rd1/Rd2 SP向后开始进行。
文库质检
文库构建完成后,先使用Qubit2.0 Fluorometer进行初步定量,稀释文库至1.5ng/ul,随后使用Agilent 2100 bioanalyzer对文库的insert size进行检测,insert size符合预期后,qRT-PCR对文库有效浓度进行准确定量(文库有效浓度高于2nM),以保证文库质量。
2.1.3 上机测序
库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行Illumina测序。测序的基本原理是边合成边测序(Sequencing by Synthesis)。在测序的flow cell中加入四种荧光标记的dNTP、DNA聚合酶以及接头引物进行扩增,在每一个测序簇延伸互补链时,每加入一个被荧光标记的dNTP就能释放出相对应的荧光,测序仪通过捕获荧光信号,并通过计算机软件将光信号转化为测序峰,从而获得待测片段的序列信息。测序过程如下图所示(图2.3):

图2.3 Illumina测序原理示意图
2.2 生信分析流程
RNA-seq的核心是基因表达差异的显著性分析,使用统计学方法,比较两个条件或多个条件下的基因表达差异,从中找出与条件相关的特异性基因,然后进一步分析这些特异性基因的生物学意义,分析过程包括质量控制、序列比对、表达水平定量、差异分析、功能富集等环节。另外可变剪接,变异位点也是RNA-seq的重要分析内容。同时,根据不同的研究需求,推出转录组个性化分析内容,如基因共表达网络构建(WGCNA)、基因集富集分析(GSEA)、ipath 代谢途径可视化分析等。信息分析流程如下图所示:
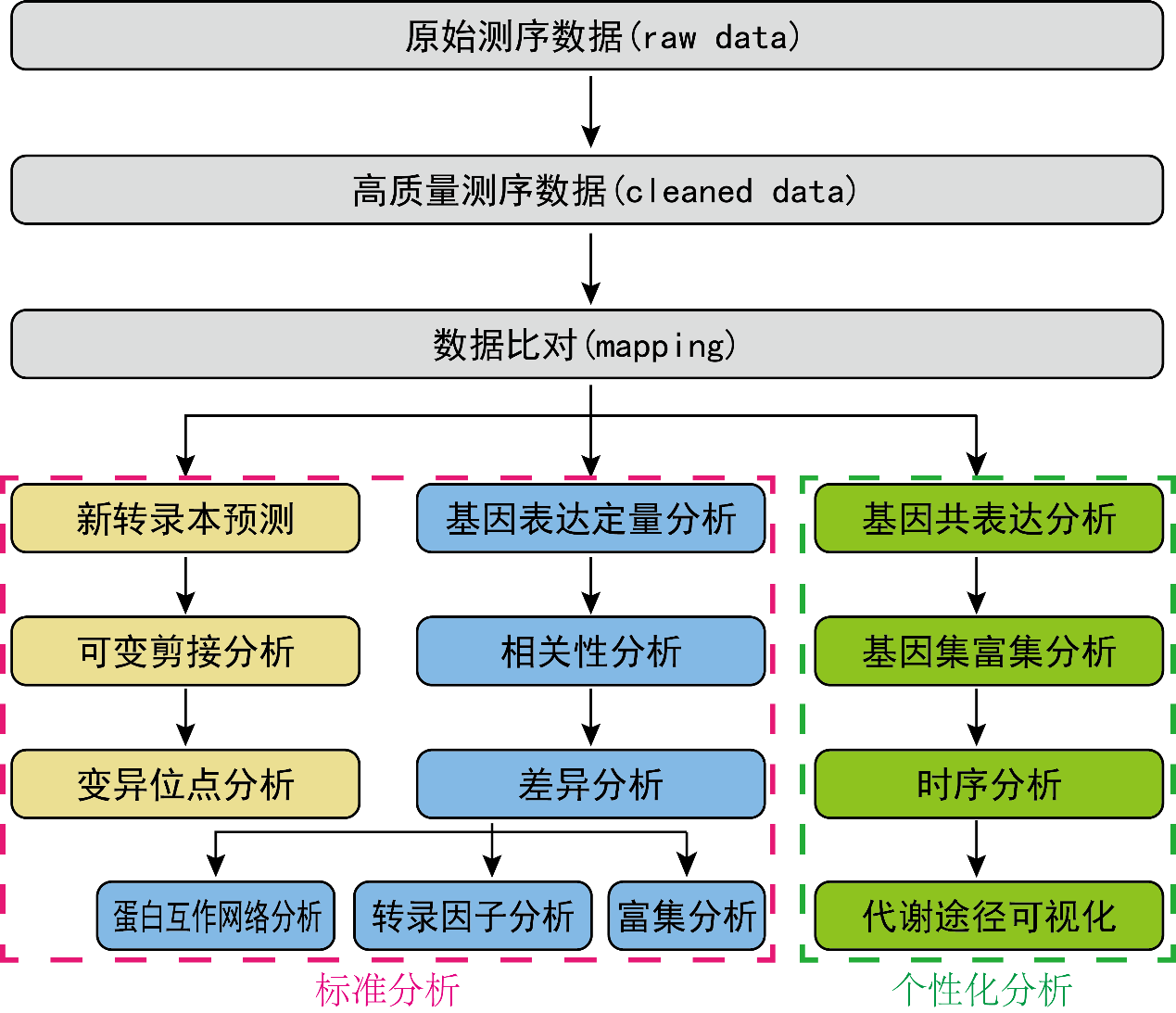
图2.4 RNA-Seq信息分析技术流程
三 结果文件夹解读
3.1 数据质量控制
01.QC/ ├── Sample*_fastp.html [各样本fastp软件质控报告] ├── summary_after_filtering.xls [各样本质控后质量情况汇总] ├── summary_before_filtering.xls [各样本质控前质量情况汇总]
用fastp软件[1]对每一个样本的测序数据raw data做质控处理(参数: --overrepresentation_analysis --trim_front1 2 --trim_front2 2 --cut_front --cut_tail --cut_window_size 3 --cut_mean_quality 30),得到clean data用于下游分析,质控规则如下:
- 对整条read做保留或弃去处理
- 根据碱基质量值弃去低质量的整条read
- 当一条read中N碱基的个数达到一定的数量(默认为5个)时,弃去整一条read;
- 当一条read中低质量碱基(默认质量值低于15的碱基为低质量碱基)达到一定的比例(默认这个比例值为40%)时,弃去整一条read。
- 根据read长度弃去低质量的整条read
- 当一条read的碱基个数少于一定的数量(默认为15个碱基)时,弃去整一条read。
- 对read做局部裁剪
- 强制性删除头部和尾部各2个碱基
- 因为测序反应一般起始和结束阶段的碱基质量是最差的,所以我们统一裁剪掉read1和read2它们各自的5'端的2个碱基、3'端的2个碱基。
- 根据碱基质量值删除滑动窗口内的碱基
- 一般来说,测出来的read,开头的碱基质量差,然后随着测序反应的稳定,碱基质量会逐步上升,后面随着反应时间的延长、化学产物的积累、酶受到的环境不利影响的积累,到后面碱基质量会下降,所以需要对read的开头和结尾的低质量碱基区做裁剪。
- 我们以3个碱基的长度为一个窗口的大小,从read的头部向尾部滑动,计算窗口内的3个碱基的平均质量值,如果平均质量值小于阈值30,那么表明当前窗口的碱基质量差,就删除这个窗口内的所有的3个碱基,然后滑动到下一组的3个碱基,计算平均质量值,做类似的删除处理;如果当前的窗口的平均质量值大于或等于30,那么表明当前窗口的碱基质量足够好了,于是停止滑动窗口,保留这个窗口以及后面的所有碱基。同理,从read的尾部向头部,也做类似的滑动窗口删除碱基的处理。
- adapter序列裁剪
- 默认情况下,对pair-end 的read1和read2做两序列比对,查看比对结果中,read1的右端是否会超出read2的右端,read2的左端是否会超出read1的左端,如果超出的话,表明是测到了adapter序列,则会把超出的部分给剪掉。
将所有样本的质控信息做提取整合,生成总体的样本测序数据质控信息统计表。
表3-1-1 各样本质控前质量情况汇总(summary_before_filtering.xls)
| sample id | total reads | total bases | total giga bases | q20 bases | q30 bases | q20 rate | q30 rate | read1 mean length | read2 mean length | gc content |
|---|---|---|---|---|---|---|---|---|---|---|
| sample06 | 48001366 | 7200204900 | 7.2G | 6985688144 | 6661033330 | 0.970207 | 0.925117 | 150 | 150 | 0.510378 |
| sample03 | 41140080 | 6171012000 | 6.17G | 5986636418 | 5702804462 | 0.970122 | 0.924128 | 150 | 150 | 0.514026 |
| sample09 | 42852266 | 6427839900 | 6.43G | 6307072859 | 6083280726 | 0.981212 | 0.946396 | 150 | 150 | 0.498650 |
| sample01 | 41048664 | 6157299600 | 6.16G | 5973286270 | 5689995335 | 0.970115 | 0.924106 | 150 | 150 | 0.514685 |
| sample07 | 42646320 | 6396948000 | 6.4G | 6274847941 | 6050079755 | 0.980913 | 0.945776 | 150 | 150 | 0.497578 |
| sample10 | 47377652 | 7106647800 | 7.11G | 6962364807 | 6706424274 | 0.979697 | 0.943683 | 150 | 150 | 0.495538 |
| sample12 | 40524798 | 6078719700 | 6.08G | 5969518231 | 5771636200 | 0.982035 | 0.949482 | 150 | 150 | 0.497519 |
| sample04 | 48224196 | 7233629400 | 7.23G | 7004083018 | 6664692845 | 0.968267 | 0.921348 | 150 | 150 | 0.514348 |
| sample08 | 41576168 | 6236425200 | 6.24G | 6118343660 | 5905132769 | 0.981066 | 0.946878 | 150 | 150 | 0.494835 |
| sample11 | 41284004 | 6192600600 | 6.19G | 6071929333 | 5856156787 | 0.980514 | 0.945670 | 150 | 150 | 0.498113 |
| sample05 | 41950854 | 6292628100 | 6.29G | 6108668503 | 5825621837 | 0.970766 | 0.925785 | 150 | 150 | 0.507869 |
| sample02 | 42098356 | 6314753400 | 6.31G | 6115892682 | 5825414218 | 0.968509 | 0.922509 | 150 | 150 | 0.508335 |
- sample id: 样本ID
- total reads: 样本序列总数
- total bases: 样本碱基总数
- total giga bases: 样本碱基总数(G为单位)
- q20 bases: 质量分高于20(错误率0.01)的碱基总数
- q30 bases: 质量分高于30(错误率0.001)的碱基总数
- q20 rate: 质量分高于20(错误率0.01)的碱基占比(Q20)
- q30 rate: 质量分高于30(错误率0.001)的碱基占比(Q30)
- read1 mean length: read1平均长度
- read2 mean length: read2平均长度
- gc content: GC含量
表3-1-2 各样本质控后质量情况汇总(summary_after_filtering.xls)
| sample id | total reads | total bases | total giga bases | q20 bases | q30 bases | q20 rate | q30 rate | read1 mean length | read2 mean length | gc content |
|---|---|---|---|---|---|---|---|---|---|---|
| sample06 | 47553720 | 6826500089 | 6.83G | 6664992959 | 6369382367 | 0.976341 | 0.933038 | 143 | 143 | 0.508873 |
| sample03 | 40806400 | 5832229700 | 5.83G | 5692377766 | 5434500770 | 0.976021 | 0.931805 | 143 | 142 | 0.512331 |
| sample09 | 42633548 | 6141035218 | 6.14G | 6047883257 | 5839294934 | 0.984831 | 0.950865 | 144 | 143 | 0.497746 |
| sample01 | 40720034 | 5820109550 | 5.82G | 5680296400 | 5422949651 | 0.975978 | 0.931761 | 143 | 142 | 0.513220 |
| sample07 | 42425864 | 6108262395 | 6.11G | 6014258122 | 5805154422 | 0.984610 | 0.950377 | 144 | 143 | 0.496586 |
| sample10 | 47099096 | 6768072718 | 6.77G | 6658663639 | 6422321809 | 0.983835 | 0.948914 | 143 | 143 | 0.494720 |
| sample12 | 40300302 | 5785914824 | 5.79G | 5705548931 | 5523232223 | 0.986110 | 0.954600 | 143 | 143 | 0.496472 |
| sample04 | 47746016 | 6789282317 | 6.79G | 6621511226 | 6316891514 | 0.975289 | 0.930421 | 142 | 141 | 0.512054 |
| sample08 | 41340254 | 5947930822 | 5.95G | 5859157455 | 5661366327 | 0.985075 | 0.951821 | 143 | 143 | 0.493983 |
| sample11 | 41039208 | 5893402563 | 5.89G | 5804048424 | 5604985201 | 0.984838 | 0.951061 | 143 | 143 | 0.497015 |
| sample05 | 41624738 | 5953089305 | 5.95G | 5813704308 | 5556414903 | 0.976586 | 0.933367 | 143 | 142 | 0.506584 |
| sample02 | 41700296 | 5983292233 | 5.98G | 5837553896 | 5573864041 | 0.975642 | 0.931571 | 143 | 143 | 0.506901 |
- sample id: 样本ID
- total reads: 样本序列总数
- total bases: 样本碱基总数
- total giga bases: 样本碱基总数(G为单位)
- q20 bases: 质量分高于20(错误率0.01)的碱基总数
- q30 bases: 质量分高于30(错误率0.001)的碱基总数
- q20 rate: 质量分高于20(错误率0.01)的碱基占比(Q20)
- q30 rate: 质量分高于30(错误率0.001)的碱基占比(Q30)
- read1 mean length: read1平均长度
- read2 mean length: read2平均长度
- gc content: GC含量
3.2 比对参考基因组
02.Mapping/
├── summary_reads_alignment.xls [各样本序列比对情况汇总]
├── summary_reads_genomic_origin.xls [各样本序列基因组比对区域分布]
├── summary_transcript_coverage_profile.xls [各样本转录本位置覆盖度统计]
└── Sample*
├── css
├── images_qualimapReport
├── qualimapReport.html [qualimap 比对质量评估报告,文件夹内其它文件都是该报告的数据,无需关注]
├── raw_data_qualimapReport
└── rnaseq_qc_results.txt
测序片段是由mRNA随机打断而来,为了确定这些片段对应基因组上的哪些区域,我们使用HISAT2软件[2]将质控后的clean data reads比对到参考基因组上(参数: --dta (为下游组装作准备)),生成.sam或.bam文件,里面包含clean data read与基因组的比对记录(alignment)。
我们随后用Qualimap RNA-seq软件[3]流程评估比对情况(参数: --paired)
表3-2-1 各样本序列比对情况汇总(summary_reads_alignment.xls)
| sample id | reads aligned (left/right) | read pairs aligned | total alignments | secondary alignments | non-unique alignments | aligned to genes | ambiguous alignments | no feature assigned | not aligned | SSP estimation (fwd/rev) | overall mapping rate |
|---|---|---|---|---|---|---|---|---|---|---|---|
| sample06 | 23,011,732 / 22,733,617 | 20,735,972 | 48,635,772 | 2,890,423 | 4,601,142 | 39,103,662 | 352,759 | 4,577,916 | 1,808,371 | 0.5 / 0.5 | 0.962 |
| sample03 | 19,745,458 / 19,495,300 | 17,454,061 | 41,768,527 | 2,527,769 | 3,996,225 | 33,662,690 | 321,407 | 3,787,980 | 1,565,642 | 0.5 / 0.5 | 0.962 |
| sample09 | 20,799,690 / 20,781,980 | 19,191,138 | 48,457,315 | 6,875,645 | 9,849,799 | 34,372,582 | 260,455 | 3,974,407 | 1,051,878 | 0.5 / 0.5 | 0.975 |
| sample01 | 19,638,224 / 19,400,291 | 17,448,415 | 41,576,869 | 2,538,354 | 4,014,499 | 33,552,491 | 319,971 | 3,689,669 | 1,681,519 | 0.5 / 0.5 | 0.959 |
| sample07 | 20,747,769 / 20,700,578 | 19,171,152 | 46,682,199 | 5,233,852 | 7,926,964 | 34,097,009 | 249,429 | 4,408,700 | 977,517 | 0.5 / 0.5 | 0.977 |
| sample10 | 22,924,073 / 22,929,526 | 21,068,745 | 54,245,244 | 8,391,645 | 11,855,082 | 37,238,999 | 295,437 | 4,855,627 | 1,245,497 | 0.5 / 0.5 | 0.974 |
| sample12 | 19,683,023 / 19,669,307 | 18,106,988 | 46,335,718 | 6,983,388 | 9,908,533 | 32,192,411 | 250,743 | 3,983,909 | 947,972 | 0.5 / 0.5 | 0.976 |
| sample04 | 23,013,618 / 22,710,420 | 19,857,866 | 48,632,676 | 2,908,638 | 4,597,542 | 39,481,459 | 370,092 | 4,183,236 | 2,021,978 | 0.5 / 0.5 | 0.958 |
| sample08 | 20,131,787 / 20,097,847 | 18,594,499 | 47,792,822 | 7,563,188 | 10,793,565 | 32,192,089 | 272,327 | 4,534,745 | 1,110,620 | 0.5 / 0.5 | 0.973 |
| sample11 | 20,030,500 / 19,999,212 | 18,400,279 | 46,209,787 | 6,180,075 | 8,931,065 | 32,832,744 | 259,983 | 4,185,917 | 1,009,496 | 0.5 / 0.5 | 0.975 |
| sample05 | 20,241,997 / 19,998,124 | 18,008,415 | 42,822,310 | 2,582,189 | 4,092,794 | 34,505,260 | 309,986 | 3,914,055 | 1,384,617 | 0.5 / 0.5 | 0.967 |
| sample02 | 19,826,623 / 19,574,453 | 18,224,337 | 42,678,299 | 3,277,223 | 5,039,281 | 33,721,525 | 439,997 | 3,477,027 | 2,299,220 | 0.5 / 0.5 | 0.945 |
- sample id: 样本ID
- reads aligned (left/right): read1/read2 比对上的总reads数,不包含secondary alignments(即次要比对, 一条read可能比对上多次,但只有一个主要比对(primary alignment),其余的都是次要比对)
- read pairs aligned: 比对上的read pairs总数,只有read1和read2同时比对上,才加1
- total alignments: 总比对数,这里是所有比对记录的总数,一条read如果比对上两次,则加2
- secondary alignments: 次要的比对记录总数,包括不唯一(有其它比对记录指向同一read)的次要比对记录总数,但不包含不唯一的主要比对,即total alignments 减去 reads aligned(read1+read2, 主要比对)
- non-unique alignments: 不唯一(有其它比对记录指向同一read)的比对记录总数,包括不唯一的主要和次要比对,但不包括唯一的主要比对,比secondary alignments稍高(多出的部分是主要比对)
- aligned to genes: 比对到基因上的reads总数
- ambiguous alignments: 比对到多个基因上(同一read)的比对记录总数
- no feature assigned: 没有比对到任何feature(如比对到基因间隔区和内含子区的比对记录)的比对记录总数
- not aligned: 未比对上的reads总数
- SSP estimation (fwd/rev): strand specificitiy proportion,可理解为比对到正链/负链的比对记录占比,对非链特异性的RNA-Seq来说,这两个占比差不多,在0.5附近
- overall mapping rate: 比对率,=reads aligned / reads cleaned = reads aligned / (reads aligned + not aligned)。如果相关实验存在细菌或其它物种的核酸片段污染,比对率可能会很低(0.1以下),但比对失败的reads是不会参与后续分析的,这相当于一个去除污染序列的过程,因此无需担心后续结果的准确性。
表3-2-2 各样本序列基因组比对区域分布(summary_reads_genomic_origin.xls)
| sample id | exonic | intronic | intergenic | overlapping exon |
|---|---|---|---|---|
| sample06 | 39,103,662 (89.52%) | 3,240,596 (7.42%) | 1,337,320 (3.06%) | 971,103 (2.22%) |
| sample03 | 33,662,690 (89.89%) | 2,688,909 (7.18%) | 1,099,071 (2.93%) | 883,622 (2.36%) |
| sample09 | 34,372,582 (89.64%) | 1,757,524 (4.58%) | 2,216,883 (5.78%) | 719,239 (1.88%) |
| sample01 | 33,552,491 (90.09%) | 2,504,852 (6.73%) | 1,184,817 (3.18%) | 846,106 (2.27%) |
| sample07 | 34,097,009 (88.55%) | 2,290,021 (5.95%) | 2,118,679 (5.5%) | 749,521 (1.95%) |
| sample10 | 37,238,999 (88.46%) | 2,327,913 (5.53%) | 2,527,714 (6%) | 825,965 (1.96%) |
| sample12 | 32,192,411 (88.99%) | 1,940,692 (5.36%) | 2,043,217 (5.65%) | 683,579 (1.89%) |
| sample04 | 39,481,459 (90.42%) | 2,939,120 (6.73%) | 1,244,116 (2.85%) | 1,081,657 (2.48%) |
| sample08 | 32,192,089 (87.65%) | 1,946,110 (5.3%) | 2,588,635 (7.05%) | 717,075 (1.95%) |
| sample11 | 32,832,744 (88.69%) | 2,078,444 (5.61%) | 2,107,473 (5.69%) | 733,524 (1.98%) |
| sample05 | 34,505,260 (89.81%) | 2,752,361 (7.16%) | 1,161,694 (3.02%) | 813,128 (2.12%) |
| sample02 | 33,721,525 (90.65%) | 2,312,544 (6.22%) | 1,164,483 (3.13%) | 885,091 (2.38%) |
- sample id: 样本ID
- exonic: 比对到外显子区的reads数(占比)
- intronic: 比对到内含子区的reads数(占比)
- intergenic: 比对到基因间区的reads数(占比)
- overlapping exon: 比对到多个外显子区的reads数(占比)
- 这里统计的比对记录不包括歧义比对(比对到多个基因),不唯一比对(比对到多个基因组位置),以及比对到未注释区域的记录
表3-2-3 各样本转录本位置覆盖度统计(summary_transcript_coverage_profile.xls)
| sample id | 5' bias | 3' bias | 5'-3' bias |
|---|---|---|---|
| sample06 | 0.64 | 0.24 | 1.61 |
| sample03 | 0.68 | 0.27 | 1.52 |
| sample09 | 0.67 | 0.33 | 1.45 |
| sample01 | 0.67 | 0.25 | 1.58 |
| sample07 | 0.65 | 0.30 | 1.39 |
| sample10 | 0.63 | 0.31 | 1.46 |
| sample12 | 0.67 | 0.31 | 1.48 |
| sample04 | 0.69 | 0.29 | 1.46 |
| sample08 | 0.56 | 0.38 | 1.41 |
| sample11 | 0.66 | 0.30 | 1.48 |
| sample05 | 0.64 | 0.22 | 1.60 |
| sample02 | 0.42 | 0.33 | 1.53 |
- sample id: 样本ID
- 5' bias: 转录本5'端平均覆盖度/整个转录本平均覆盖度
- 3' bias: 转录本3'端平均覆盖度/整个转录本平均覆盖度
- 5'-3' bias: 转录本5'端平均覆盖度/3'端平均覆盖度
- 计算每个指标时,都独立抽取基因组注释的1000条转录本进行统计,所以5' bias/3' bias不等于5'-3' bias
- 如果用poly-A selection方法去除rRNA,可以用bias来评估是否发生RNA降解,如果RNA发生降解,3'端覆盖度高,5'端低,即3' bias明显比5' bias高,5'-3' bias远小于1
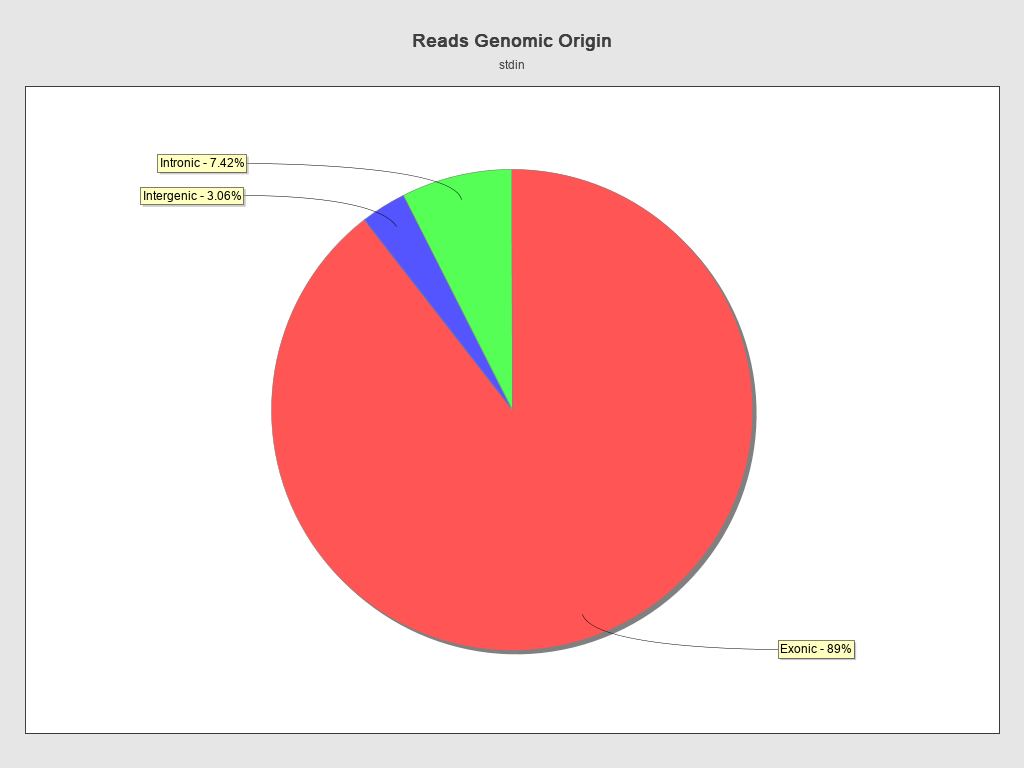
图3-2-1 某个样本序列基因组比对区域分布
该图在qualimapReport.html内
qualimapReport.html的解读请参照Qualimap RNA-seq官网
3.3 新转录本预测
03.Assemble/ ├── novel.gtf [新转录本的基因组注释文件,包含新转录本在基因组上的位置信息] ├── novel_transcripts.emapper.annotations.xls [新转录本的emapper功能注释文件, 包含新转录本在各个数据库的注释信息] └── novel_transcripts.fa [新转录本的FASTA序列文件]
新转录本预测涉及多个gtf文件的操作,gtf全称为gene transfer format,是对染色体上基因和转录本进行外显子结构注释的常用格式,其含有转录本及对应外显子的详细信息
新转录本预测步骤如下:
- 我们以每个样本的bam格式比对结果文件作为输入,使用StringTie软件[4]对每个样本单独进行转录本组装(默认参数),得到每个样本的组装转录本基因组注释(sample1.gtf, sample2.gtf...);
- 之后合并(stringtie --merge)所有转录本注释,得到一个所有样本的转录本注释(all_sample.gtf)
- 最后用gffcompare软件[5]将该all_sample.gtf与参考基因组注释文件(reference.gtf)比较(默认参数),提取参考基因组未注释过的转录本(class_code==u, 即unknown, intergenic),并筛除长度小于100以及外显子个数少于2的转录本,作为预测的新转录本(也就是novel.gtf)。
StringTie使用网络流算法以及可选的从头组装来拼接转录本。相对于cufflinks等软件,其具有以下优势:1.拼接出更完整的转录本。2.拼接出更准确的转录本。3.更好的估计转录本的表达水平。4.更快的拼接速度。
novel.gtf可用excel打开,因为表格太大,此处以图片示例展示(表3-3-1),实际以novel.gtf文件内容为准。
表3-3-1 新转录本基因组注释
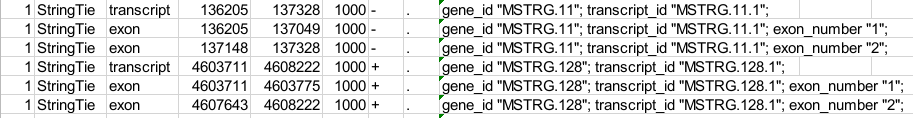
- seqname: 染色体编号
- source: 注释的来源,这里的 StringTie 是指该转录本是由 StringTie 软件组装所得
- feature: 注释信息的结构类型,如 gene、transcript、exon 等
- start: 注释区域在染色体上的起始坐标
- end: 注释区域在染色体上的终止坐标
- score: 注释信息的得分,是注释信息可能性的说明,点号表示为空
- strand: 注释区域在染色体上的正负链信息
- frame:仅对注释类型为 CDS 的有效(其它为空),有效值为 0,1,2,分别表示第一个密码子应该从start开始的第1,2,3个碱基算起
- attributes:包含众多属性的列表,主要为基因编号、转录本编号等信息。注意:有的新转录本虽然标注了参考基因ID,但其外显子结构注释与参考基因转录本外显子结构注释差异很大,故判定为新转录本
我们使用emapper软件[6]对新转录本进行了功能注释,结果见novel_transcripts.emapper.annotations.xls
3.4 变异位点分析
04.SNP/
├── all_sample.anno.vcf [vcf格式(variant call format)的所有样本的变异位点信息及其注释信息]
├── all_variant_count.xls [变异位点的计数]
└── VariantStat
├── CHROM_variant_count.svg [各染色体的变异位点计数统计图]
├── CHROM_variant_count.xls [各染色体的变异位点计数统计表]
├── Consequence_variant_count.svg [按变异后果分类的变异位点计数统计图]
├── Consequence_variant_count.xls [按变异后果分类的变异位点计数统计表]
├── IMPACT_variant_count.svg [按变异严重程度分类的变异位点计数统计图]
└── IMPACT_variant_count.xls [按变异严重程度分类的变异位点计数统计表]
这里分析的变异位点主要有两种类型: SNP和Indel
- SNP全称Single Nucleotide Polmorphisms,单核苷酸多态性。是指在基因组上由单个核苷酸变异形成的遗传标记,其数量巨大,多态性丰富。一般而言,SNP是指变异频率大于1%的单核苷酸变异。
- Indel全称Insertion deletion,小片段插入缺失。是指相对于参考基因组,样本中发生的小片段的插入缺失,该插入缺失可能含一个或者多个碱基。
变异位点分析是RNA-Seq结构分析的重要内容,主要包括先天变异位点和后天体细胞突变的检测,对肿瘤等研究具有重要意义。
我们使用GATK软件[7]对每个样本的比对数据(bam)进行变异位点分析(关键参数: gatk HaplotypeCaller --emit-ref-confidence GVCF --dont-use-soft-clipped-bases --standard-min-confidence-threshold-for-calling 30,详细过程参照官网),并使用VEP(variant effect predictor)软件[8]对变异位点进行注释(参数: --canonical --symbol --vcf --stats_text --gtf [参考基因组注释文件])。
all_sample.anno.vcf中保存了获取变异位点的过程的相关信息及变异位点的具体信息。##开头的行用于对表格内容进行注释,帮助理解表格内容,#开头的行是表头,之后的行即为表格记录,##开头的可能有几百行,需要往下拉很多才能看到表格内容。可用Excel打开,因表格过大,此处仅以实力图片展示,具体以all_sample.anno.vcf内容为准。
表3-4-1 vcf格式的所有样本的变异位点信息及其注释信息(all_sample.anno.vcf)
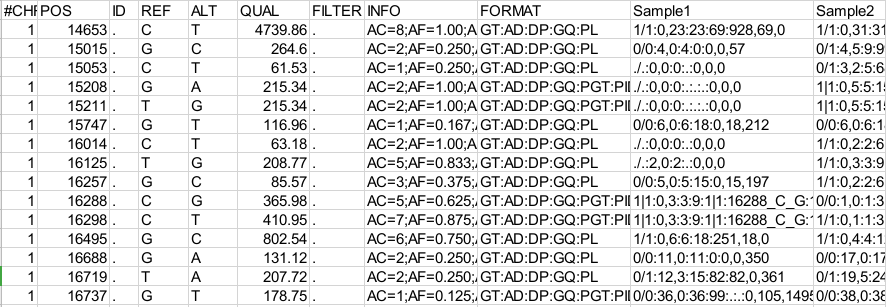
- CHROM: 变异所在染色体编号
- POS: 突变位点在染色体上的坐标
- ID: 突变ID(空)
- REF: 参考基因组的该位点的碱基
- ALT: 样本中检测到的变异碱基(对Indel来说是片段),即等位基因型,可有多个,逗号隔开
- QUAL: 变异位点质量,Phred 格式的数值,数值越高,变异位点基因型的可信度越高
- FILTER: 筛选规则(空)
- INFO: INFO有很多字段,信息量极大,";"分隔,每个字段的解释见文件头##开始的行,如“##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes, for each ALT allele, in the same order as listed">”解释了INFO里字段AC的含义(Allele count in genotypes, for each ALT allele, in the same order as listed,即所有样本检测到的对应变异总计数);这里我们主要关注最后一个字段CSQ,它即为VEP软件提供的每个变异位点的注释信息,由于一个变异位点可能对多个feature(如转录本)产生影响,这里的注释有多条记录,记录之间以逗号分隔,每条记录又有多个字段,字段之间以|分隔,各个字段从左到右分别为Allele|Consequence|IMPACT|SYMBOL|Gene|Feature_type|Feature|BIOTYPE|EXON|INTRON|HGVSc|HGVSp|cDNA_position|CDS_position|Protein_position|Amino_acids|Codons|Existing_variation|DISTANCE|STRAND|FLAGS|SYMBOL_SOURCE|HGNC_ID|CANONICAL|SOURCE|gtf,注意字段值可能为空(造成连续|),各字段含义可参考VEP官网Other output fields,需要关注的主要字段我们在all_variant_count.xls文件里列出来了,请参照下文对该文件的解释
- FORMAT: ”:“分隔的字段名,对应后续各样本列的字段值
- Sample*: ”:“分隔的字段值,对应FORMAT列的字段名;各字段的含义可以在文件开头##开头的行里找到;DP:变异位点在样本中的测序深度;AD:分别指支持 REF 与 ALT 的 reads 数,中间以逗号相隔;GT:变异位点的基因型,/分隔,二倍体有两个值,0,1,2,3....分别对应REF以及ALT里逗号分隔的多个等位基因型,也就是说,0代表没有变异
all_variant_count.xls文件保存了我们对每个变异基因型的计数,对于一个二倍体来说,它的计数只会有三个值,0,1,2,分别表示没有该变异基因型,杂合子,纯合子变异。all_variant_count.xls表格各列含义如下:
- CHROM: 变异所在染色体编号
- POS: 突变位点在染色体上的坐标
- REF: 参考基因组的该位点的碱基(基因型)
- QUAL: 变异位点质量,Phred 格式的数值,数值越高,变异位点基因型的可信度越高
- Allele: 变异的等位基因型
- SYMBOL: 变异作用的基因名
- Gene: 变异作用的基因ID
- Feature_type: 变异作用的Feature类型
- Feature: 变异作用的Feature
- BIOTYPE: 变异作用的BIOTYPE
- STRAND: 链1正链,-1负链
- Consequence: 变异后果类型
- IMPACT: 变异严重程度
- Sample*: 变异基因型的计数
我们在结果文件夹中提供了按照染色体,变异后果类型,变异严重程度分类的变异基因型的计数总和统计柱形图,图3-4-1展示了按照变异严重程度分类的变异基因型的计数统计柱形图。

图3-4-1 按变异严重程度分类的变异位点计数统计图
3.5 可变剪接分析
05.AS/
├── [分组方案]_[处理组].vs.[对照组]
│ ├── [可变剪接类型]
│ │ ├── [可变剪接类型].MATS.[JCEC|JC].Sig.txt [在分组间差异最显著的20个可变剪接,FDR<0.05]
│ │ ├── [可变剪接类型].MATS.[JCEC|JC].txt [所有可变剪接]
│ │ └── Sashimi_plot
│ │ ├── [输入文件([可变剪接类型].MATS.[JCEC|JC].Sig.txt)中的第几行数据]_[基因名]_[染色体的序号]_
[第1个外显子的起点坐标]_[第1个外显子的终点坐标]_[第1个外显子在正链还是负链]@
[其它相关外显子信息,格式同前].pdf [Sashimi Plot]
我们利用RNA-Seq数据进行可变剪接分析,可变剪接(Alternative Splicing,AS)是大多数真核生物细胞中普遍存在的转录本形式。真核生物细胞的基因序列包含内含子(intron)和外显子(exon),在基因转录成前体RNA后内含子会被剪接体移除,而外显子连接进一步加工后成为成熟的mRNA。
转录出来的前体RNA可以有多种外显子剪接形式,因此使得一个基因在不同时间、不同环境中可以翻译出不同的蛋白质,增加其生理状态下系统的复杂性和适应性。
rMATs是一款适用于RNA-Seq数据的可变剪接分析软件,它不仅可以对可变剪接事件进行分类,还可以进行不同样本间可变剪接事件的差异分析。rMATs可鉴定的可变剪接事件分类如下(图3-5-1)。

图3-5-1 rMATs可鉴定的可变剪接事件
- SE: Skipped exon 外显子跳跃
- MXE: Mutually exclusive exon 外显子互斥
- A5SS: Alternative 5' splice site 5’端外显子发生可变剪接
- A3SS: Alternative 3' splice site 3’端外显子发生可变剪接
- RI: Retained intron 内含子滞留
3.5.1 差异可变剪接事件
以每个进行差异可变剪接分析的比较组为单位,我们首先统计发生的可变剪切事件的种类及数量,然后分别计算每类可变剪切事件表达量,最后对每类可变剪切事件进行差异分析。我们使用rMATS 软件[9]进行AS 事件的定量及差异分析(参数:-t paired --readLength 100 --variable-read-length --cstat 0.0001)。
rMATS 软件可以将 AS 事件进行5 种分类,并且可以对有生物学重复的样品进行差异 AS 分析。每个可变剪接事件对应两个 Isoform,分别为 Exon InclusionIsoform 和 Exon Skipping Isoform,分别对两个 Isoform 进行表达量统计,并除以其有效长度,得到校正后的表达量,然后计算 Exon Inclusion Isoform 在两个Isoform 总表达量的比值,比值即为结果文件中的 IncLevel1 (处理组)和 IncLevel2(对照组),如下图所示。最后进行差异显著性分析 。
[可变剪接类型].MATS.[JCEC|JC].Sig.txt文件里包含了在分组间差异最显著的20个可变剪接(FDR<0.05),可用excel打开,各列含义如下:
- ID: 可变剪接ID
- GeneID: 可变剪接事件所在基因编号
- geneSymbol: 可变剪接事件所在基因名称
- chr: 可变剪接事件所在染色体
- strand: 可变剪接事件所在染色体链的方向
- exonStart_0base: 可变剪接事件跳跃外显子起始位置,以 0 开始计数
- exonEnd: 可变剪接事件跳跃外显子终止位置
- upstreamES: 可变剪接事件跳跃外显子的上游 exon 起始位置
- upstreamEE: 可变剪接事件跳跃外显子的上游 exon 终止位置
- downstreamES: 可变剪接事件跳跃外显子的下游 exon 起始位置
- downstreamEE: 可变剪接事件跳跃外显子的下游 exon 终止位置
- ID.1: 与ID一样
- IJC_SAMPLE_1: 可变剪接事件 Exon Inclusion Isoform 在差异比较组合中处理组的表达量
- SJC_SAMPLE_1: 可变剪接事件 Exon Skipping Isoform 在差异比较组合中处理组的表达量
- IJC_SAMPLE_2: 可变剪接事件 Exon Inclusion Isoform 在差异比较组合中对照组的表达量
- SJC_SAMPLE_2: 可变剪接事件 Exon Skipping Isoform 在差异比较组合中对照组的表达量
- IncFormLen: 可变剪接事件 Exon Inclusion Isoform 的有效长度
- SkipFormLen: 可变剪接事件 Exon Skipping Isoform 的有效长度
- PValue: 可变剪接事件表达差异显著性 p 值
- FDR: 可变剪接事件表达差异显著性 FDR 值
- IncLevel1: 处理组可变剪接事件 Exon Inclusion Isoform 在两个 Isoform 总表达量的比值
- IncLevel2: 对照组可变剪接事件 Exon Inclusion Isoform 在两个 Isoform 总表达量的比值
- IncLevelDifference: IncLevel1 与 IncLevel2 的差值
“[可变剪接类型].MATS.[JCEC|JC].Sig.txt” 与 ”[可变剪接类型].MATS.[JCEC|JC].txt” 格式一致
3.5.2 差异剪接可视化
对于表达差异显著性的可变剪接事件, 我们用rmats2sashimiplot软件[10]进行可视化展示,SE 类型可变剪接事件的可视化展示如下图所示:图中标题为可变剪接事件三个外显子所在的染色体坐标及正负链信息,跨外显子比对的 reads 使用连接外显子junction 边界的弧线表示。弧线的粗细和比对到 junction 上的 reads 数成正比,同时弧线上的数字指出了 junction reads 的数目,右上方标注了各个样本可变剪接事件所在的基因及 IncLevel 值,横坐标为 AS 发生在染色体上的位置,纵坐标表示发生该可变剪切需要的 reads 数。
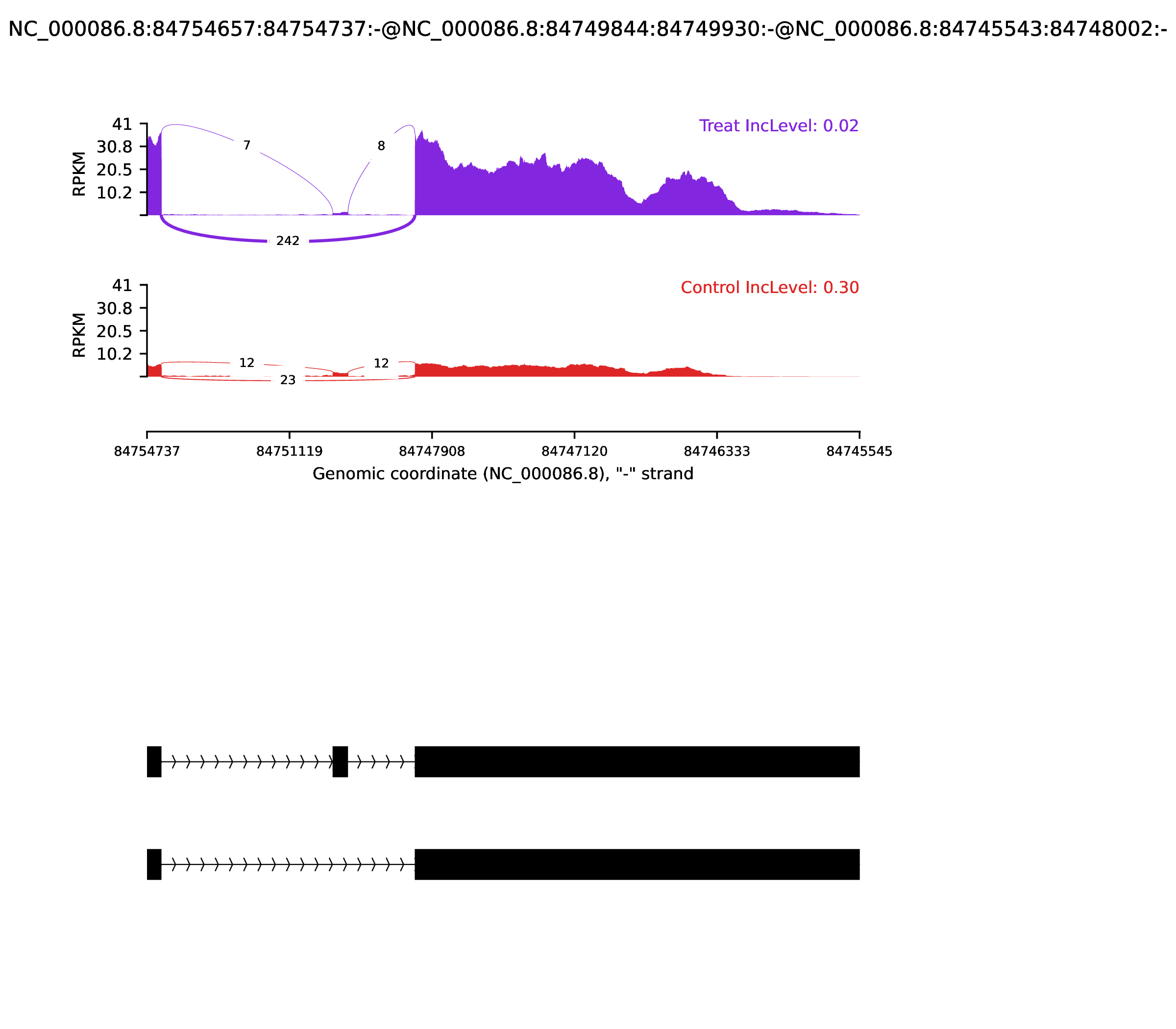
图3-5-2 SE 类型可变剪接可视化展示(Sashimi_plot)
图中红色和橘黄色分别表示不同的样本,inclusion level 值代表 exon Inclusion Isoform 在两个 Isoform总表达量的比值,可以直接看出不同样本中可变剪接事件的表达差异
3.6 定量分析
06.Quant/
├── CountsQC [qualimap Counts QC报告]
│ ├── [样本]Report.html [单个样本的报告]
│ ├── GlobalReport.html [所有样本的总报告]
| └── * [报告数据和图片]
├── gene_count.xls [基因比对到的reads数矩阵]
├── gene_deseq2.xls [用DESeq2的方法校正后的表达量矩阵]
├── gene_edger_TMM.xls [用edgeR的方法校正后的表达量矩阵]
├── gene_fpkm.xls [FPKM表达量矩阵]
├── gene_tpm.xls [TPM表达量矩阵]
├── boxplot
│ ├── Category_boxplot.svg [TPM表达量分布箱线图]
│ └── Category_data_matrix.xls [箱线图数据]
├── heatmap
│ ├── [分组方案]_correlation_heatmap.svg [TPM表达量样本间spearman相关性热图]
│ ├── [分组方案]_correlation_matrix.xls [TPM表达量样本间spearman相关系数矩阵]
│ ├── [分组方案]_p_value_matrix.xls [TPM表达量样本间spearman相关系数p值矩阵]
├── pca
│ ├── [分组方案]_PCA_3D.svg [TPM表达量3D PCA图]
│ ├── [分组方案]_pca_points_ordinates.xls [PCA各样本坐标]
│ └── [分组方案]_PCA.svg [TPM表达量PCA图]
└── violin
├── [分组方案]_data_matrix.xls [小提琴图数据]
└── [分组方案]_violin.svg [TPM表达量分布小提琴图]
用subRead软件包中的featureCounts软件[11]计算gene count(gene_count.xls), 关键计算参数为: -p -t exon -g gene_id。这里的count指的是fragment count, 即一对reads是按照一个整体(fragment)来计数的。
FPKM和TPM的计算请参照文献中的公式,这两个表达量可用于比较样本或分组之间的表达量差异,也能用于比较基因之间的表达量差异
以gene_count.xls为输入,筛除检出率(count不为0的比例)小于0.25的基因,用R语言DESeq2包[12]和edgeR包[13]分别计算校正后的表达量gene_deseq2.xls和gene_edger_TMM.xls;这两个表达量表格可用于比较样本或分组之间的表达量差异,但不能用于比较基因之间的表达量差异。请注意,这个两个文件并不能作为DESeq2和edgeR的直接输入文件,也不能用于计算DESeq2和edgeR的FoldChange,DESeq2和edgeR只能用gene_count.xls作为输入文件,因为它们已经将校正过程编码到模型中
3.6.1 测序reads饱和度分析
我们用qualimap软件[3]对gene fragment count文件做了counts QC分析,其中测序reads饱和度Saturation分析,可以用来大致评估每一个样本的测序量是否能充分覆盖有表达的基因。
qualimap countsQC软件的底层使用R语言NOIseq包,关于qualimap Counts QC报告的解读可以查看 NOIseq官网文档 以及 qualimap官网
此处展示所有样本的测序reads饱和度分析曲线:
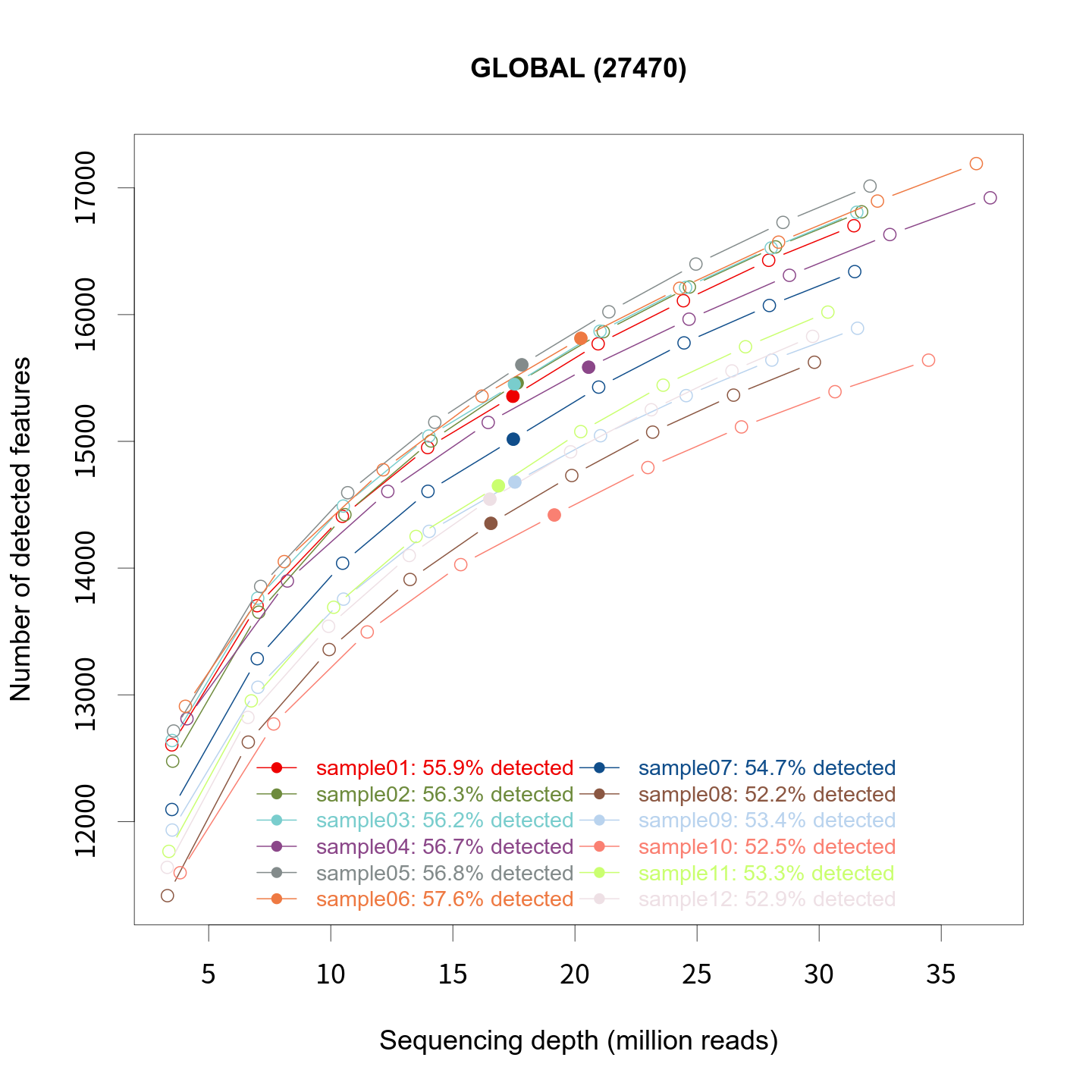
图3-6-1 测序reads饱和度分析曲线
- 图中正上方的GLOBAL右边括号中的数字,表示整个gene fragment count表格文件中的数据记录的行数。
- 横轴表示有多少pair的reads比对到基因组上,纵轴表示有多少种基因被识别到。测序量充足的情况下,当测序reads达到一定数量后,能把明显有表达的基因给识别出来,所以再增加测序reads的话,识别到的基因的种类数不会明显增多,曲线趋于平缓。
- Saturation图中,曲线中的画实心的圆点,其横坐标表示该样本的实际的所有的reads pair数,其纵坐标表示用该样本的实际的所有read能检测到的基因的种类数,一个基因上要超过5个pair reads落在上面才算是检测到了该基因。在实心圆点左侧以及右侧的空心圆点都是根据gene fragment count表格文件模拟出来的数据点。图中下方的数字含义是,某个样本实际检测到的有fragment count值的基因,占整个gene fragment count表格中的基因的种类数的比例,例如,整个gene fragment count表格中有50000个数据行,我们说有50000种不同的基因在这个表格中,而某个样本中,大于5的fragment count的数据行有10000个,我们说它实际检测到了10000种基因,那么计算10000/50000 = 20%,得到这个值。
3.6.2 基因表达分布
为展示各个样本的基因表达量的整体的分布情况,我们对所有样本的所有基因的TPM值,用R语言程序作“小提琴图”和“箱线图”。为避免图中数据点的跨度太大,我们对TPM值,做了log2(TPM+1)的缩小变换处理。
箱线图可以展示样本基因表达量分布情况:

图3-6-2 基因表达分布箱线图
箱子下,中,上沿分别对应,下四分位点,中位数,上四分位点,竖线标明了±3倍标准差的范围
小提琴图也可用于展示样本基因表达量分布情况:

图3-6-3 基因表达分布小提琴图
小提图琴是个密度图,越宽的地方说明对应于纵轴表达量的基因越多。
3.6.3 样本间相关性
用样本的TPM或FPKM数据计算两两样本间的相关性系数,通过观察相关性系数的大小,可以大致评估样本组内的生物学实验重复性如何。数据只保留了在本分组方案中,在所有的样本中的TPM之和大于0的基因,而把完全不表达的基因的信息给略去了。
Encyclopedia of DNA Elements (ENCODE) Consortium,是由美国国家人类基因组研究所(NHGRI)资助的,一个国际合作研究小组。ENCODE的目标是构建人类基因组功能元件的信息,包括在蛋白质和RNA水平上起作用的元件,以及控制细胞和基因活动环境的调控元件。
ENCODE project 2016年的RNA-Seq指导方案中写到,样本间相关系数要在0.9 或0.8以上:
- 对于来自同一个人体供体(或者同一个模式生物菌株)的生物学重复样本,要求在0.9以上;
- 对于来自不同的人体供体(或者模式生物不同的菌株)的相近的组织的生物学重复样本,要求在0.8以上。

图3-6-4 TPM表达量样本间spearman相关性热图
热图中的单元格颜色越深,表示对应的两个样本之间的相关性系数越高。同一个小组内的样本应该具有较高的相关性系数。同一个小组内的样本应该具有较高的相关性系数。
3.6.4 主成分分析
主成分分析(PCA)也常用来评估组间差异及组内样本重复情况,PCA采用线性代数的计算方法,对数以万计的基因变量进行降维及主成分提取。我们对所有样本的基因表达值(TPM)进行PCA分析,如下图所示。理想条件下,PCA图中,组间样本应该分散,组内样本应该聚在一起

图3-6-5 主成分分析结果图
图中横坐标为第一主成分,纵坐标为第二主成分,不同的颜色表示不同的分组。样本点之间的距离近似于样本之间各基因TPM差异的总和
3.7 挑选差异表达基因
07.DEG/
├── [分组方案]_[处理组].vs.[对照组]
│ ├── [分组方案]_[处理组].vs.[对照组]_All_KEGG.xls [KEGG基因差异分析的完整结果,用于下游KEGG GSEA分析]
│ ├── [分组方案]_[处理组].vs.[对照组]_All.xls [基因差异分析的完整结果,用于下游GO GSEA分析]
│ ├── [分组方案]_[处理组].vs.[对照组]_DEG_KEGG.xls [显著差异的KEGG基因集,用于下游KEGG ORA分析]
│ ├── [分组方案]_[处理组].vs.[对照组]_DEG_STRING.xls [显著差异的STRING基因集,用于下游PPI(蛋白互作网络)分析]
│ ├── [分组方案]_[处理组].vs.[对照组]_DEG.xls [显著差异的基因集,用于下游GO ORA分析,Venn|upset图,聚类热图]
│ ├── [分组方案]_[处理组].vs.[对照组]_Volcano.svg [差异分析火山图]
│ ├── heatmap.svg [差异最显著的基因表达量聚类热图]
│ ├── input.xls [聚类热图输入表达量矩阵]
│ └── table.xls [聚类热图最终数据]
└── VennUpSet
├── DEG_upset.svg [各差异基因集之间的UpSet图]
├── DEG_upset_venn_intersects.xls [各差异基因集之间的交集]
├── DEG_venn.svg [各差异基因集之间的Venn图]
├── summary_up_down.svg [各差异基因集上调,下调数目汇总柱形图]
└── summary_up_down.xls [各差异基因集上调,下调数目汇总表]
在转录组中,确定某个基因在不同的样品中的表达量是否有差异是分析的核 心内容之一。获得基因表达量后,即可对表达数据进行统计学分析,进而筛选不 同样本之间显著差异的基因。差异分析主要分为四个步骤:
- 以gene_count.xls为输入,筛除检出率(count不为0的比例)小于0.25的基因,这些基因通常不服从分析模型的数据分布假设,因而不适合做差异分析
- 对count进行标准化(normalization),主要是对测序深度的 校正。
- 通过统计学模型进行假设检验,计算p value
- 进行多重假设检验校正(BH方法),获得经校正的p value (结果表中的padj)
针对不同的实验情况,我们选用合适的软件进行基因表达差异显著性分析, 具体如下表所示:
表3-7-1 表达差异分析所用软件
| 类型 | R语言包 | 标准化方法 | 数据分布假设 | pvalue计算模型 | padj计算方法 |
|---|---|---|---|---|---|
| 有生物学重复 | DESeq2[12] | DESeq2 | 负二项分布 | 广义线性模型 | BH |
| 无生物学重复 | edgeR[13] | TMM | 负二项分布 | 广义线性模型 | BH |
一般来说,如果一个基因在两组样品中的表达量差异达到两倍以上,我们认 为这样的基因是具有表达差异的。为了判断两个样品之间的表达量差异究竟是由 于各种误差导致的还是本质差异,我们需要对所有基因在这两个样本中的表达量 数据进行假设检验。而转录组分析是针对成千上万个基因进行的,这样会导致假 阳性的累积,基因数目越多,假设检验的假阳性累积程度会越高,所以引入 padj 对假设检验的 P-value 进行校正,从而控制假阳性的比例 。
差异基因的筛选标准是非常重要的,我们给出的标准|log 2 (FoldChange)| > 1 & padj< 0.05 是常用的经验值,也是我们分析流程的默认值。在实际项目中可以根据情况灵活选择,例如,差 异倍数可以选择 1.5 倍,也可以选择 3 倍,padj 常用的阈值包括 0.01、0.05、0.1 等。若按照以上标准筛选得到的差异基因过少,很有可能导致后面的功能富集分 析没有显著性结果。若项目实验只关注某几个基因的表达情况(如基因敲除), 不在意富集结果,从下面的差异分析表格中筛选关注的那几个基因即可。反之, 如果得到的差异基因数目过多,不利于后续目标基因的筛选,这个时候可使用更 严格的阈值标准进行筛选。生科云的云流程提供了调整筛选阈值的途径。
结果文件夹中,[分组方案]_[处理组].vs.[对照组]_*.xls表格文件的各字段解释如下
- Features: 基因ID
- log2FoldChange: 处理组与对照组基因表达水平的比值,再经过差异分析软件收缩模型处理,最后以 2 为底取对数
- logCPM: edgeR结果特有,log2(所有样本校正后表达量均值)
- LR: edgeR结果特有,检验统计量,用于计算p值,无需关注
- baseMean: DESeq2结果特有,所有样本校正后表达量均值
- pvalue: 显著性检验的p值
- padj: BH方法校正后的p值
- -log10(pvalue): -log10(pvalue)
- -log10(padj): -log10(padj)
- Gene Symbol: 基因名
- KEGG Gene ID: KEGG数据库对应的基因ID
- GO: GO数据库对应的基因本体条目
- STRING Protein: STRING数据库对应的蛋白ID
- TF Family: 基因转录因子家族注释
- description: 基因功能描述
火山图同时展示了差异分析的-log10(pvalue)和log2FoldChange

图3-7-1 差异分析火山图
标出基因名的是差异显著且padj最小的基因
summary_up_down.svg汇总了各差异基因集上调(处理组表达量高于对照组),下调数目

图3-7-2 各差异基因集上调,下调数目汇总柱形图
如果有多个比较方案,多个差异基因集,我们将用Venn图和UpSet图(DEG_venn.svg|图3-7-3, DEG_upset.svg|图3-7-4)展示各个差异基因集之间的交集大小(共有的差异基因数目),以及各个差异基因集特有的差异基因。Venn图和UpSet图表达的信息是一样的,只是用了不同的数据可视化形式。Venn图最多能展示五个差异基因集的关系,而UpSet图能展示的差异基因集个数能够更多。

图3-7-3 各差异基因集之间的Venn图

图3-7-4 各差异基因集之间的UpSet图
差异基因聚类热图(heatmap.svg,图3-7-5)展示了最显著的(padj最小)的基因的表达量矩阵。为了与p值计算方法保持一致,对于没有生物学重复的比较方案,我们用6.Quant/gene_edger_TMM.xls作为输入,有生物学重复则用6.Quant/gene_deseq2.xls。为了保证图片的可读性,我们展示的基因不会超过20个(云流程可调整)

图3-7-5 差异最显著的基因聚类热图
图中用颜色代表表达量,数据跨度太大会导致差异无法区分,我们对表达量进行标准化处理(减去平均值,除以标准差);聚类到一起的样本表达模式比较接近
转录因子的注释
如果提供了AnimalTFDB(v3.0)数据库(动物)或者PlantIFDB数据库(植物)中的物种,我们会在差异分析结果表格([分组方案]_[处理组].vs.[对照组]_*.xls)中增加基因的"TF Family"注释结果。
转录因子(transcription factor)是一类蛋白质,它们能与某些目标基因的5'端上游的特定DNA序列专一性结合,从而使得目标基因以特定的表达量在特定的时间与空间(生物体内的某些组织)表达。数据库对每个转录因子家族都做了详细的介绍,请参考AnimalTFDB(v3.0)数据库(动物)或者PlantIFDB数据库(植物),理解每个转录因子家族。
3.8 富集分析
08.Enrich
├── GSEA
│ ├── GO
│ │ └── [分组方案]_[处理组].vs.[对照组]
│ │ ├── circular_cnetplot.svg [富集分析p值(p.adjust)最小的条目的环状基因概念网络]
│ │ ├── cnetplot.svg [富集分析p值(p.adjust)最小的条目的基因概念网络]
│ │ ├── dotplot.svg [富集分析p值(p.adjust)最小的条目的气泡图]
│ │ ├── emapplot.svg [富集分析p值(p.adjust)最小的条目的emapplot]
│ │ ├── enrich.xls [富集分析结果表格]
│ │ ├── heatplot.svg [富集分析p值(p.adjust)最小的条目的基因概念瀑布图]
│ │ ├── summary_gseaplot.svg [富集分析p值(p.adjust)最小的条目的gseaplot]
│ │ └── treeplot.svg [富集分析p值(p.adjust)最小的条目的聚类树图]
│ └── KEGG
│ └── [分组方案]_[处理组].vs.[对照组]
│ ├── circular_cnetplot.svg
│ ├── cnetplot.svg
│ ├── dotplot.svg
│ ├── emapplot.svg
│ ├── enrich.xls
│ ├── heatplot.svg
│ ├── KEGG_pathview [包含富集分析p值(p.adjust)最小的KEGG通路的通路图的文件夹]
│ ├── summary_gseaplot.svg
│ └── treeplot.svg
└── ORA
├── GO
│ └── [分组方案]_[处理组].vs.[对照组]
│ ├── circular_cnetplot.svg
│ ├── cnetplot.svg
│ ├── dotplot.svg
│ ├── emapplot.svg
│ ├── enrich.xls
│ ├── heatplot.svg
│ └── treeplot.svg
└── KEGG
└── [分组方案]_[处理组].vs.[对照组]
├── circular_cnetplot.svg
├── cnetplot.svg
├── dotplot.svg
├── emapplot.svg
├── enrich.xls
├── heatplot.svg
├── KEGG_pathview
└── treeplot.svg
注: 同名文件解释相同,不赘述
本报告所有富集分析使用的都是R语言clusterProfiler包[14]。
关于本报告中使用的富集分析的详细介绍,请参考clusterProfiler包教程
对于KEGG富集分析,参考基因组基因结构注释文件中使用的基因ID,和KEGG数据库使用基因ID 是不一样的,在进行KEGG通路富集前,我们需要将参考基因组基因ID转换为KEGG数据库使用的基因ID。如果KEGG数据库存在该物种基因ID与参考基因组基因ID的对应关系,我们可以直接匹配。但大部分非模式物种不存在这种匹配关系,在这种情况下,我们就用emapper软件通过序列比对寻找KEGG数据库的直系同源基因组(KEGG Orthology, K+数字,如K00010),用于KEGG富集分析。无论哪种ID转换方式,都有相当一部分基因无法找到匹配记录,从而无法参与富集分析
GO富集分析的情况也类似。在进行GO富集分析之前,我们需要知道每个基因属于哪些GO条目。当NCBI数据库存在该物种的基因与GO条目的对应关系时,我们可以直接匹配。但大部分非模式物种不存在这种匹配关系,在这种情况下,我们就用emapper软件通过序列比对寻找匹配的GO条目
为了保证结果图的可读性,cnetplot.svg, heatplot.svg, gseaplot.svg图选取最显著的(p.adjust最小)5个条目作图。dotplot.svg, treeplot.svg, emapplot.svg图选取最显著的20个条目作图。云流程可自由调整
ORA分析
在上游分析得到了组间表达量有显著性差异的基因数据后,我们可以在基因注释文件中查到单个基因的功能。此外,我们还可以探究这些具有研究意义的基因中,是否显著地对应着一些功能分类的基因的集合,从而推断差异表达基因主要参与的生物学过程
KEGG代谢通路数据库中,有很多前人归纳的生物的代谢通路,一个代谢通路包含了一定数量的基因,那么我们得到的差异表达的基因集合中,会不会存在一种情况,即大量的基因集中出现在一个或几个代谢通路中,如果是这样,那么不同处理所影响的生物学过程就更加明确了。类似的,常见的功能富集分析还有GO条目富集分析。
ORA分析结果表格(ORA/[KEGG|GO]/enrich.xls)各列含义如下
- ID: 富集到的条目(如KEGG通路)的ID
- Description: 富集到的条目(如KEGG通路)的描述
- GeneRatio: 匹配到该条目的差异基因数/差异基因总数
- BgRatio: 该条目一共有多少基因/所有条目一共有多少基因
- pvalue: 显著性检验p值
- p.adjust: BH方法校正后的p值
- qvalue: 校正错误发现率(FDR)的p值
- geneID: 与该条目相匹配的差异基因ID或基因名
- Count: 匹配到该条目的差异基因数
围绕该结果表格,我们做了很多可视化。
GO ORA

图3-8-1 富集分析p值(p.adjust)最小的条目的气泡图(dotplot.svg)
点大小对应Count: 匹配到该条目的差异基因数,即enrich.xls表Count列(ORA结果)或者core_enrichment列基因个数(GSEA结果);颜色对应p.adjust; BH方法校正后的p值; 横坐标对应GeneRatio: 匹配到该条目的差异基因数/差异基因总数

图3-8-2 富集分析p值(p.adjust)最小的条目的基因概念网(cnetplot.svg)
图中,每一簇的节点的中心是GO条目,这些外围的节点是对应的基因(即enrich.xls表geneID列(ORA分析)或者core_enrichment列(GSEA分析)),有一些基因可能会同时拥有多种GO条目的功能描述。 图中右侧的图例,size表示某一个GO条目对应有多少个基因(即enrich.xls表Count列(ORA结果)或者core_enrichment列基因个数(GSEA结果)),在作图区中表现为,一个GO条目对应的基因个数越多,则该GO条目的圆点越大;而所有的基因节点的面积大小是相同的,这些节点没有表征个数多少的含义; 图中右侧的图例,fold change,它指的是上游的差异分析中,得到的在某个比较组合中每一个基因的log2FoldChange值,作图区中的基因的节点的颜色跟log2FoldChange值是直接对应的;而中心的GO条目圆点的颜色都是相同的,它们的颜色不表征log2FoldChange值的含义; 给线条涂了不同的颜色,使得不同的GO条目延伸出来的线条颜色是不同的;

图3-8-3 络富集分析p值(p.adjust)最小的条目的环状基因概念网络(circular_cnetplot.svg)
circular_cnetplot.svg是前面的cnetplot.svg的一种特殊画法,图中元素的内容本质上是一样的, 只是把所有的基因节点与GO条目圆点都尽量画在一个圆周上。

图3-8-4 富集分析p值(p.adjust)最小的条目的基因概念瀑布图(heatplot.svg)
这个热图是非聚类热图,表达的意思和cnetplot.svg相似。横轴代表基因,按照英文字母从A到Z的顺序,把基因从左到右排列;纵轴代表GO条目,按照英文字母从A到Z的顺序,把GO条目从下往上排列。 图中右侧的图例,fold change,它指的是上游的差异分析中,得到的在某个比较组合中每一个基因的log2FoldChange值,作图区中的基因对应的单元格的颜色跟log2FoldChange值是有关联的。由于log2FoldChange只跟基因有关,而与GO条目无关,所以在横轴的某一个基因的上方,如果出现了多个有颜色的单元格,那么它们的颜色应该是相同的;而如果在某个位置没有出现涂色的单元格,则表示enrich.xls表中在纵轴坐标对应的GO条目的geneID列(ORA分析)或者core_enrichment列(GSEA分析)中不包含横轴对应的这个基因。
KEGG ORA
除上文介绍的富集结果图外,KEGG ORA在KEGG_pathview文件夹中还有最显著的通路图。
图3-8-5 富集分析p值(p.adjust)最小的KEGG通路的通路图
图中红 色表示上调基因,绿色表示下调基因。鼠标悬停于标记节点,可弹出差异基因的 细节信息,主要包括基因 ID, log2FoldChange和 padj 值。 点击各个节点,可以连接到 KEGG 官方数据库中各个节点的具体信息页。
GSEA分析
ORA分析关注的是差异表达基因,然而可能筛不到差异表达基因。GSEA可以用所有基因参与分析,GSEA在一个基因集(如某个GO条目或者KEGG通路)中累加计算一个统计量,因此可以用于检测一些协同作用的小效应,这很重要,因为很多表型差异是由很多协同作用的小效应造成的。
给定一个预先定义的基因集S(如一个GO条目的成员基因集),和一个依据某个指标排好序的基因列表L(本报告中L采用所有基因的log2FoldChange排序),GSEA的目的在于检验S的成员是随机分散在L中(p.adjust>0.05),还是主要集中在L的底部或者顶部(p.adjust<0.05); 在本报告中,enrich.xls中的p.adjust<0.05也就意味着对应富集条目(如某个GO条目或者KEGG通路)的大部分成员基因低表达,或者高表达
除上文介绍的富集结果图外,GSEA分析还有一个专门为其量身打造的GSEA图
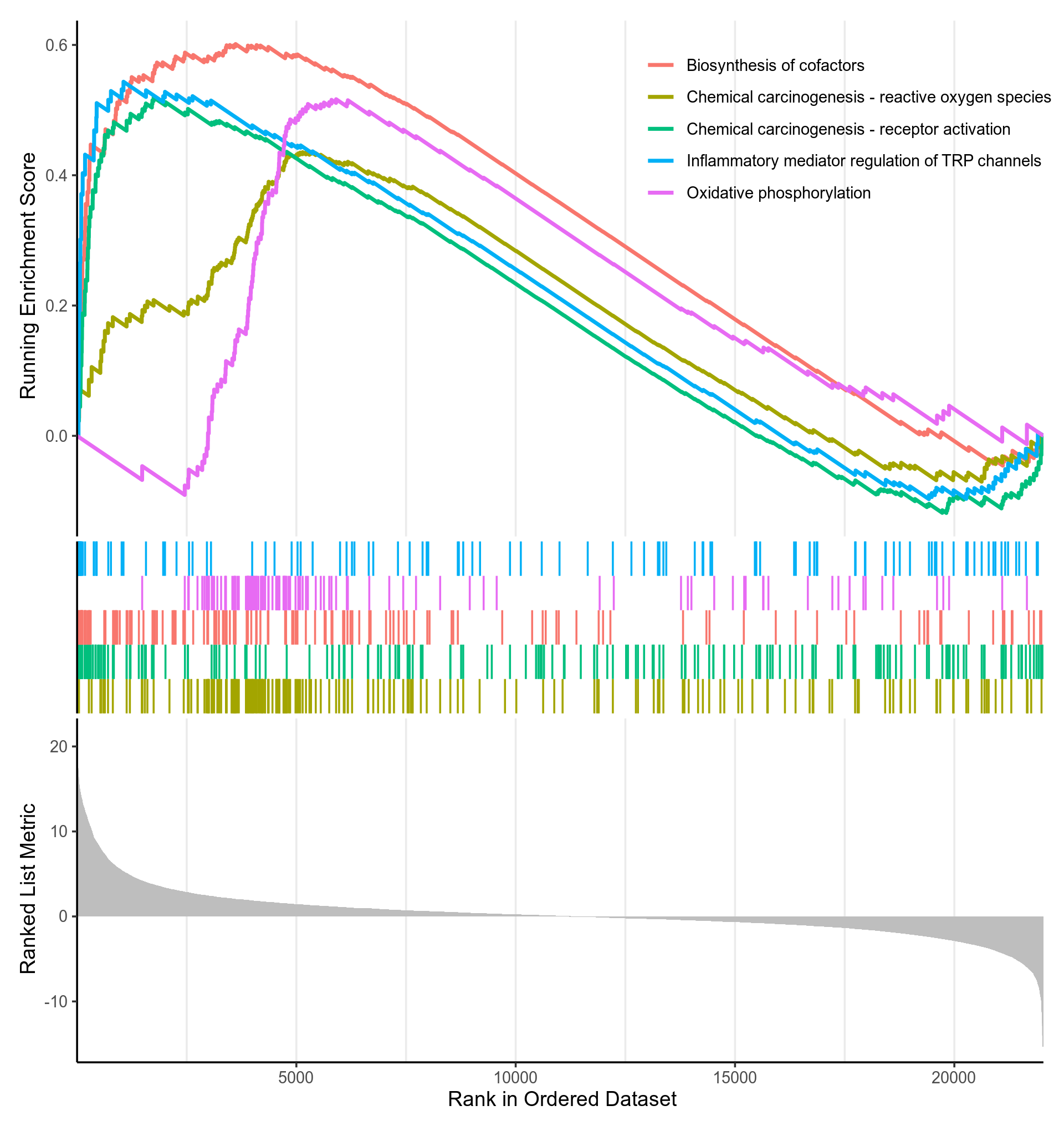
图3-8-6 GSEA富集分析p值(p.adjust)最小的KEGG通路的gseaplot
- 该图分为三部分。横坐标都代表排好序的基因列表L,如1代表log2FoldChange最高的基因。
- 对于上部分:纵坐标Running Enrichment Score的计算方式如下:从基因集L的第一个基因开始,计算一个累计统计值。当遇到一个落在S里面的基因,则增加统计值。遇到一个不在S里面的基因,则降低统计值。如果S的成员是随机分散在L中,则对应基因集的线(某个KEGG通路的线,一种颜色代表一个KEGG通路)应该是平缓的。如果存在一个单峰,则表示大部分S的成员聚集在峰的区域(不包含峰后面的部分),该区域的基因即enrich.xls的core_enrichment列(S的子集),该单峰的纵坐标即为富集得分(enrich.xls的enrichmentScore),横坐标即为enrich.xls的rank列
- 中间部分用竖线代表S的成员(某个KEGG通路的成员),有竖线,代表S包含该基因(KEGG通路包含该基因),竖线密集的地方就是Running Enrichment Score峰值出现的部位,每种颜色代表一个单独的S(KEGG通路)
- 下部分展示了排好序的基因列表L的排序指标,这里的纵坐标就是log2FoldChange
GSEA分析的enrich.xls的各列解释如下:
- ID: 富集到的条目(如KEGG通路)的ID
- Description: 富集到的条目(如KEGG通路)的描述
- setSize: 富集到的条目(如KEGG通路)的所有成员基因个数
- enrichmentScore: 富集分数
- NES: 标准化富集分数,GSEA的关键统计量,用于计算p值(置换检验)
- pvalue: 显著性检验p值(置换检验)
- p.adjust: 校正后p值(BH方法)
- qvalues: 校正后p值(FDR方法)
- rank: Running Enrichment Score达到最高值的排序位置(图3-8-6对应峰值的横坐标)
- leading_edge: 这些指标是用来表征core_enrichment(S的子基因集)的算法度量,无需关注。实在好奇,请参考GSEA官方解释
- core_enrichment: 核心富集基因集,即enrichmentScore峰值往前的S的子基因集
语义相似性分析
富集结果中的emapplot.svg和treeplot.svg是富集条目之间语义相似性分析结果图,这里使用的相似性指数是Jaccard correlation coefficient,计算公式如下:
即两个富集条目所包含基因集合[enrich.xls表geneID列(ORA分析)或者core_enrichment列(GSEA分析)]的交集基因数除以并集基因数

图3-8-7 富集分析p值(p.adjust)最小的条目的emapplot(emapplot.svg)
- 节点的大小对应enrich.xls的Count列(ORA结果)或者core_enrichment列基因个数(GSEA结果)
- 节点的颜色对应enrich.xls的p.adjust列
- 节点之间的连线代表Jaccard correlation coefficient。可理解为连线越粗,两个条目共有的基因个数(标准化后)越多

图3-8-8 富集分析p值(p.adjust)最小的条目的聚类树图(treeplot.svg)
- 节点的大小对应enrich.xls的Count列(ORA结果)或者core_enrichment列基因个数(GSEA结果)
- 节点的颜色对应enrich.xls的p.adjust列
- 这是一个依据条目之间Jaccard correlation coefficient进行聚类的树图,聚到一起的条目之间的Jaccard correlation coefficient较大,即共有基因数较多。每种颜色代表一个聚类,从聚类中随机挑选一个单词(通常是聚类中词频比较高的单词)作为该聚类的名称写在右边。
3.9 蛋白互作网络分析
09.PPI/ ├── [分组方案]_[处理组].vs.[对照组] │ ├── edges.xls [网络边(连线)数据] │ ├── network.html [差异基因的蛋白互作网络图,可动态调整网络] │ └── nodes.xls [网络节点(蛋白)数据]
该分析主要是从STRING数据库[15]搜索差异表达基因对应蛋白的互作网络,用网络图展示。
进行该分析时,我们需要将参考基因组的基因ID与STRING数据库的蛋白ID匹配,如果STRING数据库存在对应物种的ID匹配关系,我们可以直接匹配。但大部分非模式物种不存在这种匹配关系,这时候,我们用emapper软件寻找参考基因组的蛋白的直系同源蛋白簇(COGs, KOGs),用于蛋白互作网络分析
为了保证网络的可读性,我们对网络做如下筛选
- 初步筛选可信度(combined_score)最高的前1000个互作关系
- 最终筛选连接度(degree)最高的5个节点(关键基因|蛋白),以及与之互作的节点
这些筛选阈值可在云流程调整
图3-9-1 差异基因的蛋白互作网络图
为了提升交互体验,可打开一个新的网页操作网络
网络边(连线)数据(edges.xls)各列解释如下:
- protein1: 某个互作关系的第一个蛋白ID
- protein2: 某个互作关系的第二个蛋白ID
- database: 数据库注释的蛋白互作可信度
- database_transferred: 从其他物种(依据同源性)推断的database
- experiments: 实验验证的蛋白互作可信度
- experiments_transferred: 从其他物种(依据同源性)推断的experiments
- coexpression: 共表达可信度
- coexpression_transferred: 从其他物种(依据同源性)推断的共表达可信度
- textmining: 文献报道的蛋白互作可信度
- textmining_transferred: 从其他物种(依据同源性)推断的textmining
- fusion: 存在基因融合的可信度
- neighborhood: 染色体上距离较近的可信度
- neighborhood_transferred: 从其他物种(依据同源性)推断的neighborhood
- cooccurence: 共现性可信度
- combined_score: 综合计算后蛋白互作可信度
网络节点(蛋白)数据(nodes.xls)各列解释如下:
- nodes: 互作的节点ID,对应edges.xls的protein1和protein2
- Gene ID: 基因ID
- Gene Symbol: 基因名
- preferred_name: STRING数据库提供的节点名
- protein_size: 蛋白分子量
- annotation: 蛋白描述
- log2FoldChange: 差异分析中的log2FoldChange
- -log10(padj): 差异分析中的-log10(padj)
- up_down: 差异分析中是上调(up)还是下调(down)
- absolute(log2FoldChange): log2FoldChange绝对值
3.10 WGCNA分析
10.WGCNA/
├── 1.Modules [鉴定模块文件夹]
│ ├── [模块名]_genes_KEGG.xls [某个模块内的KEGG基因,用于下游KEGG富集分析]
│ ├── [模块名]_genes.xls [某个模块内的基因,用于下游GO富集分析]
│ ├── input_batch_effect_removed.xls [去除批次效应后的输入文件]
│ ├── mean_connectivity.svg [去除批次效应后的输入文件]
│ ├── model_fit.svg [不同power(幂次)下拓扑模型的拟合度]
│ ├── modules_dendrogram_power[模型所用power].svg [模块基因聚类树]
│ └── modules_ME.xls [Module eigengene E矩阵(可理解为模块表达量)]
├── 2.Network
│ ├── edges.xls [拓扑重叠网络边数据]
│ ├── network.html [交互式拓扑重叠网络]
│ └── nodes.xls [拓扑重叠网络节点数据]
├── 3.Enrich
│ ├── GO
│ │ ├── [模块名] [对应模块GO富集分析(ORA)结果]
│ │ └── *
│ └── KEGG
│ ├── [模块名] [对应模块KEGG富集分析(ORA)结果]
│ └── *
└── 4.Correlation
├── correlation_heatmap.svg [模块与表型的相关性热图]
├── correlation_matrix.xls [模块与表型的相关系数(pearson)矩阵]
└── pvalue_matrix.xls [模块与表型的相关系数p值矩阵]
加权基因共表达网络分析 (WGCNA, Weighted correlation network analysis)是用来描述不同样品之间基因关联模式的系统生物学方法,可以用来鉴定高度协同变化的基因集, 并根据基因集的内连性和基因集与表型之间的关联鉴定候补生物标记基因或治疗靶点。
相比于只关注差异表达的基因,WGCNA利用全部基因的信息识别感兴趣的基因集,并与表型进行显著性关联分析。一是充分利用了信息,二是把数千个基因与表型的关联转换为数个基因集与表型的关联,免去了多重假设检验校正的问题。
共表达网络定义为加权基因网络。点代表基因,边代表基因表达相关性。加权是指对相关性值进行幂次运算(幂次的值也就是软阈值 (power, model_fit.svg展示了确定合适的power的过程))。无符号(unsigned)网络的边属性计算方式为abs(cor(genex, geney)) ^ power;有符号(signed)网络的边属性计算方式为(1+cor(genex, geney)/2) ^ power; sign hybrid的边属性计算方式为cor(genex, geney)^power if cor>0 else 0。这种处理方式强化了强相关,弱化了弱相关或负相关,使得相关性数值更符合无标度网络特征,更具有生物意义。
WGCNA分析将共表达网络转换为TOM拓扑重叠矩阵 (Topological overlap matrix),以降低噪音和假相关,获得的新距离矩阵。当两个基因在共表达网络中拥有较多相同的"邻居"时,我们说这两个基因拓扑重叠
WGCNA分析利用TOM拓扑重叠矩阵对基因进行聚类,将拓扑重叠的基因聚为一个模块。Module(模块)指的是高度內连的基因集。在无符号(unsigned)网络中,模块内是高度相关的基因。在有符号网络中,模块内是高度正相关的基因。把基因聚类成模块后,我们对每个模块进行两个层次的分析:1. 功能富集分析查看其功能特征是否与研究目的相符;2. 模块与性状进行关联分析,找出与关注性状相关度最高的模块
鉴定模块
WGCNA相关分析使用R语言WGCNA包[16]完成。我们默认以gene_deseq2.xls作为输入文件,计算pearson相关系数,用有符号(signed)的方式加权以构建共表达网络,,进而鉴定模块。
图3-10-1展示了不同power(幂次)下,WGCNA拓扑模型的拟合度,拟合度绝对值高于0.85时的power作为最终加权幂次(所选power在文件名modules_dendrogram_power%s.svg中)。

图3-10-1 不同power(幂次)下拓扑模型的拟合度
图3-10-2展示了不同power(幂次)下,加权共表达网络中基因的平均连接度。连接度太高,不利于模块的区分,可能导致最终结果里只有一个模块,发生这种现象的原因主要有三个:
- 有离群样本
- 各分组间样本差异太大(批次效应)
- 样本数太少。样本少时,基因之间倾向于有较高的相关系数值。举个极端例子,当只有两个样本时,任意两个基因之间的相关系数都是1,无论怎么分,只可能有一个模块。通常样本数多于15个时,才能有较好的模块鉴定结果。

图3-10-2 不同power(幂次)下基因平均连接度
modules_ME.xls文件中给出了每个模块的基因成员(最后一列),以及它在各个样本的Module eigengene E,它是模块第一主成分的值,本质为各个基因表达量的加权和,可以把它理解为这个模块的表达量。
[模块名]_genes.xls文件中:
- _MM结尾的列: 表示对应模块的模块隶属度(Module Membership, MM), 即基因与该模块的相关系数
- _MM_p结尾的列: 表示对应MM的p值(默认使用holm方法校正)
- _GS结尾的列: 表示基因性状显著性(Gene Significance, GS), 即基因与该性状(环境因子)的相关系数
- _GS_p结尾的列: 表示对应GS的p值(默认使用holm方法校正)
图3-10-3 展示了各个模块基因根据TOM拓扑重叠矩阵聚类的结果。只有高度差(近似'1-拓扑重叠指数'得到的距离)在高度阈值(默认0.5,云流程可调整)以内的基因,才有可能被归为一个模块。grey是无法归类到任何模块的基因的集合
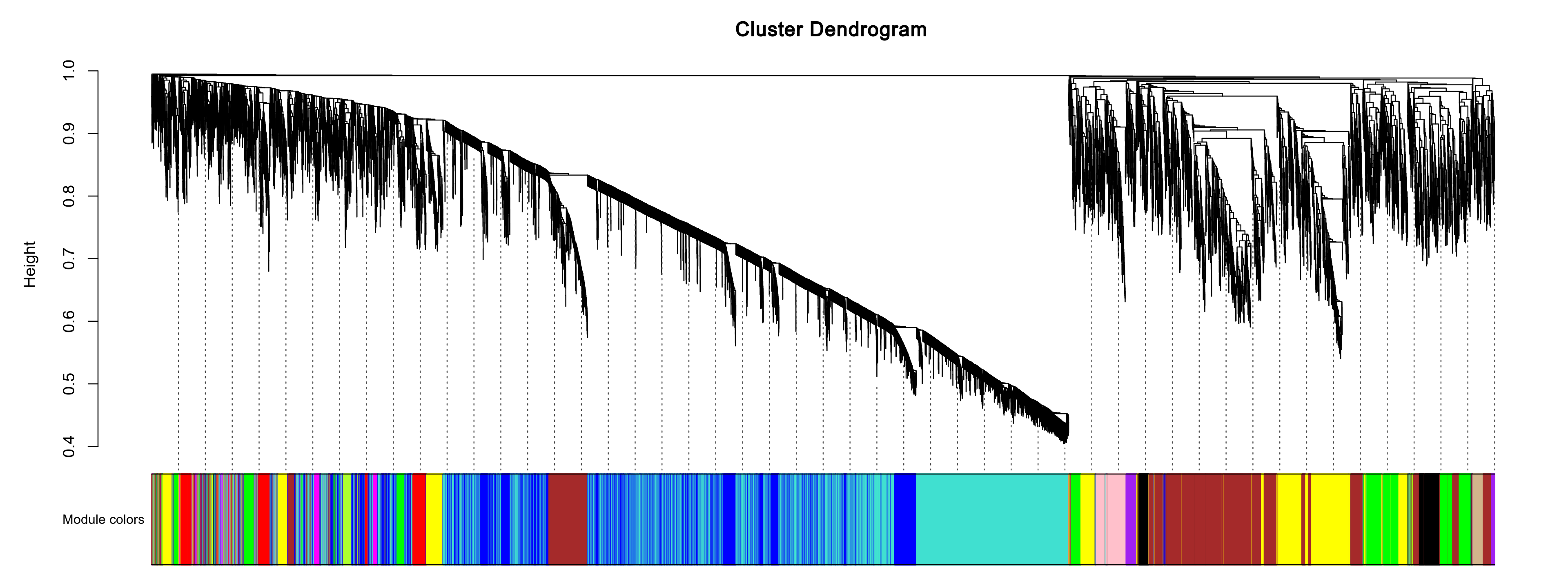
图3-10-3 模块基因聚类树
上方聚类树是所有基因根据TOM拓扑重叠矩阵聚类产生的树, 下方用不同颜色标注了不同模块。灰色基因是无法归类到任何模块的基因的集合
模块拓扑重叠网络
我们用网络图展示每个模块的基因的拓扑重叠指数,模块内相连的基因有较高的拓扑重叠指数
考虑到无标度网络的特征(连接度高的节点极少,连接度低的节点较多),同时也为了网络的可读性,对于每个模块,我们首先挑选拓扑重叠指数最高的1000个连接,计算每个节点的连接度(连线数量),最后挑选连接度最高的1个节点,以及与之相连的节点,作网络图(3-10-4)。这些挑选阈值可在云流程调整
图3-10-4 模块拓扑重叠网络
图片需要1~5分钟来加载,请耐心等候。为了提升交互体验,可打开一个新的网页操作网络
模块功能富集
为了研究每个模块主要参与哪些生物学过程,我们对每个模块进行了KEGG通路富集分析(ORA)以及GO条目富集分析。富集分析结果的解读请参照上文对富集分析结果的描述。模块富集到的功能,可以用来推断模块的功能。
模块关联表型
当老师提供表型数据(至少两个连续的数值型变量时),我们可以将模块的Module eigengene E(modules_ME.xls)与表型数据做相关分析,从而推断模块影响的表型

图3-10-5 模块与表型的相关性热图
格子的颜色对应相关系数(pearson)的大小,格子内上方标注了相关系数(pearson), 下方标注了相关系数的p值
四 分析所用软件的版本
| 软件 | 版本 |
|---|---|
| fastp | 0.23.1 |
| HISAT2 | 2.2.1 |
| QualiMap | v.2.2.2-dev |
| StringTie | 2.1.5 |
| gffcompare | v0.12.6 |
| emapper | 2.0.1 |
| GATK | v4.2.2.0 |
| VEP | 104.3 |
| rMATS | v4.1.1 |
| rmats2sashimiplot | none |
| featureCounts | v2.0.1 |
| DESeq2 | 1.26.0 |
| edgeR | 3.28.1 |
| clusterProfiler | 3.14.3 |
| WGCNA | 1.70.3 |