微科盟真核无参转录组分析结题报告
一、 概述
转录组是指特定组织或细胞在某个时间或某个状态下转录出来的所有RNA的总和, 主要包括mRNA和非编码RNA。由于自然界物种繁多且一些生物的基因组组装困难,目前我 们无法获取所有生物的完整基因组。使用有参转录组的分析策略来分析参考基因组组装较差 的物种,会导致序列比对率低,许多基因无法被检测到,从而浪费大量的数据。真核无参转 录组测序,就是针对无参考基因组或参考基因组组装较差物种的一种较好的分析策略。
真核无参转录组测序采用Illumina测序平台,对真核生物的所有RNA进行测序, 根据高质量数据进行拼接,将拼接得到的最长转录本unigene作为参考序列,进一步进行表 达定量、功能注释、SNP及SSR等分析。转录组研究是基因功能及结构研究的基础和出发点, 真核无参转录组测序为无参考基因组物种的转录水平变化、分子机制及调控网络研究提供了 有力的技术手段,目前已广泛应用于基础研究、临床诊断、药物研发和分子育种等领域。
二、 建库测序
从RNA样品提取到最终数据获得,样品检测、建库、测序等每一环节都会直接影响数据的数量和质量,从而影响后续信息分析的结果。为从源头保证测序数据准确可靠,承诺在数据的所有生产环节都严格把关,从根源上确保高质量数据的产出。建库测序的流程图如下:
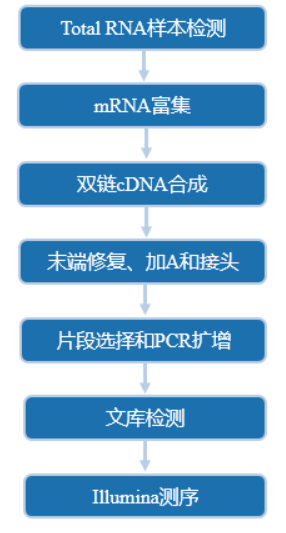
图2-1 建库测序流程
2.1 RNA提取与检测
采用标准提取方法从组织或细胞中提取RNA,随后对RNA样品进行严格质控,质控标准如下。
(1) 琼脂糖凝胶电泳分析RNA降解程度以及是否有污染
(2) Nanodrop检测RNA的纯度(OD260/280比值)
(3) Qubit对RNA浓度进行精确定量
(4) Agilent 2100精确检测RNA的完整性
注:以RNA完整性和总量为主要参考指标。
2.2 文库构建与质检
2.2.1 文库构建
mRNA的获取主要有两种方式:一是利用真核生物大部分mRNA都带有polyA尾的结构特征, 通过Oligo(dT)磁珠富集带有polyA尾的mRNA。二是从总RNA中去除核糖体RNA,从而 得到mRNA。随后在NEB Fragmentation Buffer中用二价阳离子将得到的mRNA随机 打断,按照NEB普通建库方式或链特异性建库方式进行建库。
NEB普通建库:以片段化的mRNA为模版,随机寡核苷酸为引物,在M-MuLV逆转录酶体系 中合成cDNA第一条链,随后用RNaseH降解RNA链,并在DNA polymerase I 体系下, 以dNTPs为原料合成cDNA第二条链。纯化后的双链cDNA经过末端修复、加A尾并连接测 序接头,用AMPure XP beads筛选250-300bp左右的cDNA,进行PCR扩增并再次使用 AMPure XP beads纯化PCR产物,最终获得文库。建库原理如下面左图所示。
链特异性建库:逆转录合成cDNA第一条链方法与NEB普通建库方法相同,不同之处在于合 成第二条链时,dNTPs中的dTTP由dUTP取代,之后同样进行cDNA末端修复、加A尾、连 接测序接头和长度筛选,然后先使用USER酶降解含U的cDNA第二链再进行PCR扩增并获得 文库。链特异性文库具有诸多优势,如相同数据量下可获取更多有效信息;能获得更精准 的基因定量、定位与注释信息;能提供反义转录本及每一isoform中单一exon的表达水平。 建库原理如下面右图所示。
在真核生物建库时,默认采用NEB普通建库方式进行建库。
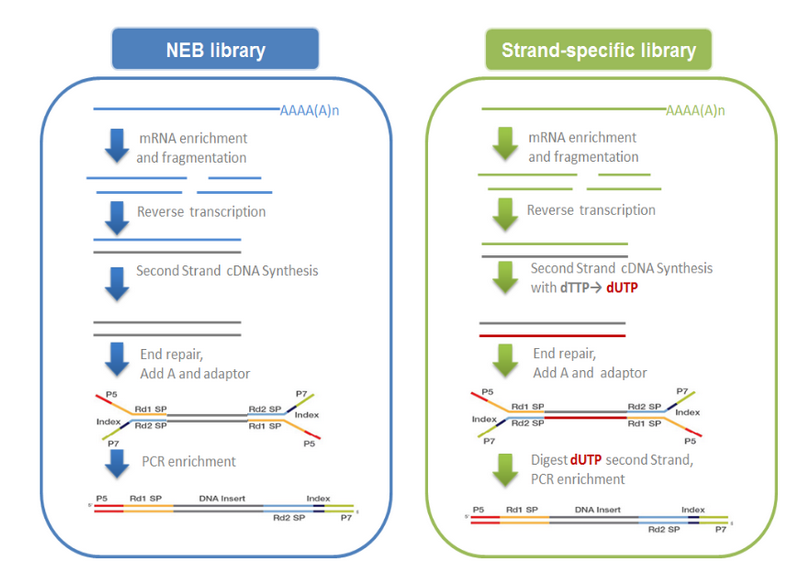
图2-2 文库构建原理示意图
注:测序接头:包括P5/P7,index和Rd1/Rd2 SP三个部分 (如上图所示)。其中P5/P7是PCR扩增引物及flow cell上引物结合的部分, index提供区分不同文库的信息,Rd1/Rd2 SP即read1/read2 sequence primer ,是测序引物结合区域,测序过程理论上由Rd1/Rd2 SP向后开始进行。
2.2.2 文库质检
文库构建完成后,先使用Qubit2.0 Fluorometer进行初步定量,稀释文库至 1.5ng/ul,随后使用Agilent 2100 bioanalyzer对文库的insert size 进行检测,insert size符合预期后,qRT-PCR对文库有效浓度进行准确定量 (文库有效浓度高于2nM),以保证文库质量。
2.3 上机测序
库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行 Illumina测序。测序的基本原理是边合成边测序(Sequencing by Synthesis)。 在测序的flow cell中加入四种荧光标记的dNTP、DNA聚合酶以及接头引物进行扩增, 在每一个测序簇延伸互补链时,每加入一个被荧光标记的dNTP就能释放出相对应的荧光, 测序仪通过捕获荧光信号,并通过计算机软件将光信号转化为测序峰,从而获得待测片段 的序列信息。测序过程如下图所示:

图2-3 Illumina测序原理示意图
三、 信息分析流程
对于无参考基因组的转录组分析,可先将测序所得的序列拼接成转录本,用Corset[3]程序 对转录本进行层次聚类,以聚类后序列为参考,进行后续的分析,过程包括质控、七大数据 库注释(Nr、Nt、KOG、Swiss-prot、Uniprot、KEGG和GO)、定量、差异显著性分析 、功能富集等环节,另外变异位点,SSR也是无参转录组的分析内容。 信息分析流程图如下:
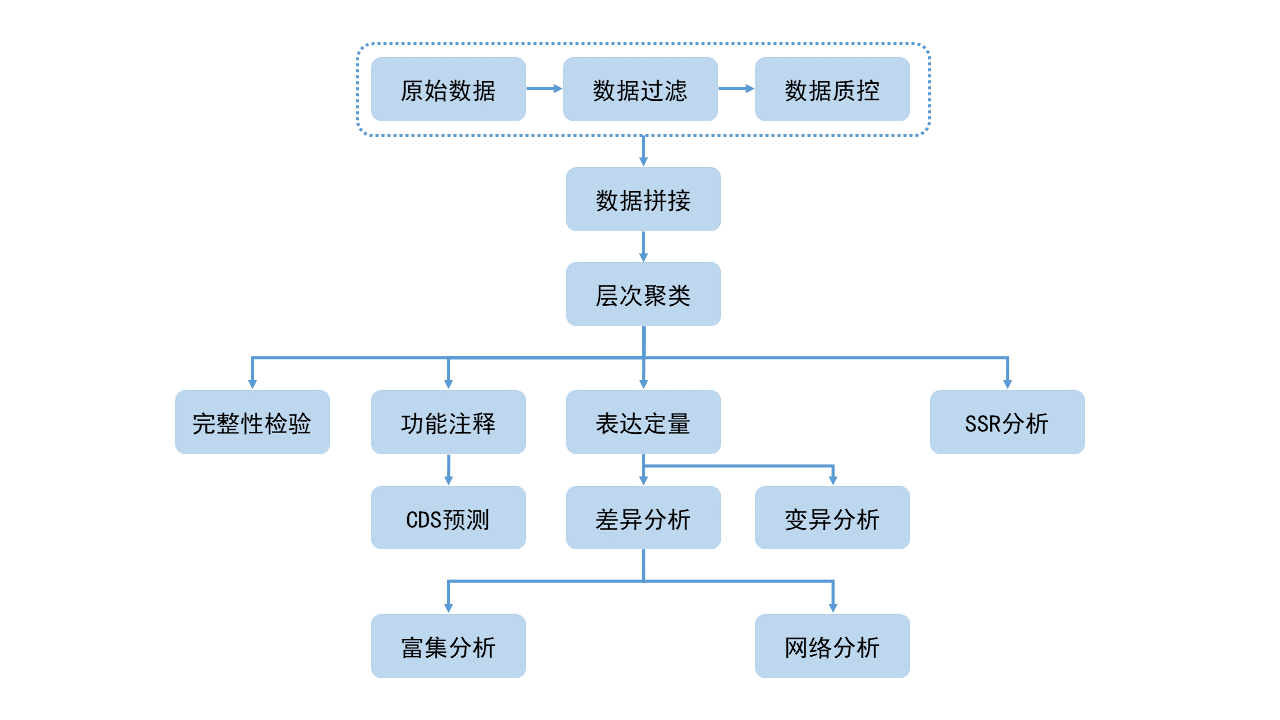
图3-1 分析流程图
3.1 质控分析
3.1.1 测序数据说明
测序片段被高通量测序仪测得的图像数据经CASAVA碱基识别转化为序列数据(reads), 文件为fastq格式,其中主要包含测序片段的序列信息以及其对应的测序质量信息。
fastq格式文件中每个read由四行描述信息组成,如下所示:

上述文件中第一行以“@”开头,随后为Illumina测序标识符(Squence Identifiers) 和描述文字;第二行是测序片段的碱基序列;第三行以“+”开头,随后为Illumina测序 标识符(也可为空);第四行是测序片段每个碱基相对应的测序质量值,该行每个字符对 应的ASCII值减去33,即为该碱基的测序质量值。
3.1.2 测序数据过滤
测序获得的原始数据中包含少量带有测序接头或测序质量较低的reads,为保证数据分 析的质量及可靠性,需要对原始数据进行过滤。本分析使用Trimmomatic[1]软件对 测序数据进行过滤,过滤前后各部分reads所占比例均在饼图中呈现。本项目测序数 据过滤情况统计图见结果文件 customer_files/01Trimmomatic。下图为其中一个样本作为示例:

图3-2 测序数据过滤情况
注:
Both Surviving:该部分为过滤后read1和read2均被保留的数据
Forward Only Surviving:该部分为仅在read1中被保留的数据
Reverse Only Surviving:该部分为仅在read2中被保留的数据
Dropped:该部分为因接头或数据质量等原因被丢弃的数据
3.1.3 测序错误率分布
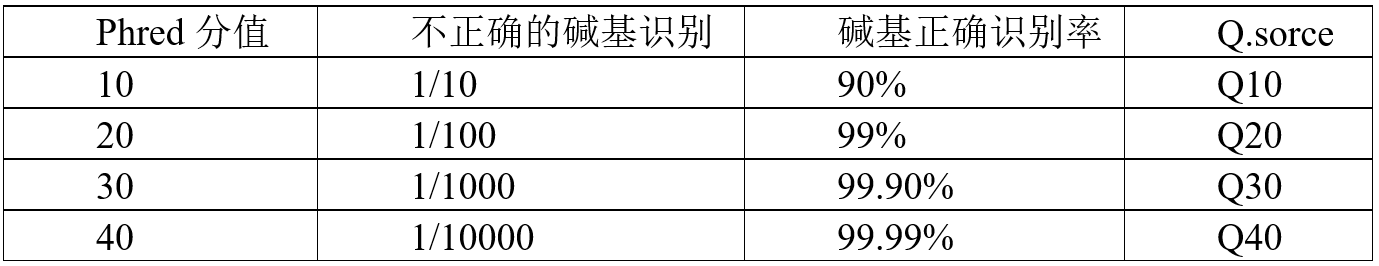
测序过程本身存在机器错误的可能性,测序错误率分布检查可以反映测序数据的质量, 序列信息中每个碱基的测序质量值保存在fastq文件中。如果测序错误率用e表示, Illumina的碱基质量值用Qphred表示,则有:Qphred=-10log10(e)。 Illumina Casava 1.8版本碱基识别与Phred分值之间的简明对应关系见下表。
表3.1 Illumina Casava 1.8版本碱基识别与Phred分值之间的简明对应关系
当前RNA-seq测序技术,测序错误率分布存在以下两个特征。
测序错误率随着测序序列(Sequenced Reads)长度的增加而升高。 这是由测序过程中化学试剂的消耗导致的,为Illumina高通量测序平台所具有的特征。
前6个碱基具有较高的测序错误率,此长度恰好为RNA-seq建库过程中反转录所需的随机引物长度。 前6个碱基测序错误率较高是因为随机引物和RNA模版的不完全结合。 此特征为illumina高通量测序平台的共有特征。
在该部分分析中,若样品80%的测序序列错误率在0.1%以下即为合格。 本项目测序数据的错误分布图见结果文件 customer_files/02QC/02.01ErrorRate/。 下图为其中一个样本作为示例:

图3-3 测序错误率分布
注:
pos:横坐标为reads碱基位置,其中从0-150为read1碱基位置,151-300为read2碱基位置。
errorRatio:纵坐标为碱基错误率。
3.1.4 GC含量分布
核苷酸序列中鸟嘌呤(G)和胞嘧啶(C)所占的比例称为GC含量。 GC含量在物种间存在一定特异性,但由于反转录过程中所使用的6bp随机引物, 会引起前几位碱基在核苷酸组成上有一定偏好性,产生正常波动,随后则趋于稳定。 对于NEB普通建库方法,由于序列的随机性打断和双链互补等原则, 理论上测序读段在每个位置的GC及AT含量应分别相等,且在整个测序过程基本稳定不变, 呈水平线。而对于链特异性建库而言,由于只保留了单链信息, 可能会出现AT分离或GC分离现象。
本项目各个样本的GC含量分布见结果文件 /customer_files/02QC/02.02GCContent。 下图为其中一个样本作为示例:

图3-4 样本GC含量分布
注:
base position:横坐标为reads碱基位置;
base count:纵坐标为该碱基所占比例。
3.1.5测序数据质量分布
测序数据的质量主要分布在 Q30(≥80%)以上,这样才能保证后续分析的正常进行, 根据测序技术的特点,测序片段末端的碱基质量一般会比前端的低。
本项目各个样本测序数据质量分布见结果文件 customer_files/02QC/02.03QualityDistribution。 下图为其中一个样本作为示例:

图3-5 样本测序质量分布
注:
pos:横坐标为碱基位置;
quality:碱基的测序质量值。
下表为数据情况一览表,其实际数据表格在文件夹 customer_files/02QC中
表3.2 测序数据一览表
| sample id | raw bases(G) | raw sequences | clean bases(G) | clean sequences | q20(%) | q30(%) | GC(%) | error_rate(%) |
|---|---|---|---|---|---|---|---|---|
| high1 | 0.75 | 5000000 | 0.07 | 476842 | 100.0 | 99.91 | 42.0(%) | 0.03 |
| high2 | 0.75 | 5000000 | 0.07 | 476208 | 100.0 | 99.92 | 43.0(%) | 0.03 |
| low1 | 0.75 | 5000000 | 0.07 | 476328 | 100.0 | 99.9 | 43.0(%) | 0.03 |
| low2 | 0.75 | 5000000 | 0.07 | 478084 | 100.0 | 99.91 | 43.0(%) | 0.03 |
| middle1 | 0.75 | 5000000 | 0.07 | 476880 | 100.0 | 99.92 | 43.0(%) | 0.03 |
| middle2 | 0.75 | 5000000 | 0.07 | 475912 | 100.0 | 99.9 | 43.0(%) | 0.03 |
注:
sample id:样本ID;
raw bases(G):样本原始碱基数量;
raw sequences:样本原始碱基序列数量;
clean bases(G):样本过滤后碱基数量;
clean sequences:样本过滤后序列数量;
q20(%):样本测序质量值超过20的碱基含量;
q30(%):样本测序质量值超过30的碱基含量;
GC(%):样本过滤后碱基GC含量;
error_rate(%):样本平均测序错误率。
3.2 转录本拼接
对于无参考基因组的项目,获得clean reads后,需要对clean reads进行拼接 以获取后续分析的参考序列。采用Trinity[2]对clean reads进行拼接。 为了识别个别样品中低表达或者只测到部分片段的基因,对于同物种的所有样品 进行合并组装,可以使组装结果更全面,并有利于差异分析等后续分析;而对于 不同物种的样品,由于基因组间差异较大,适合分别组装。
3.2.1 拼接过程
对于无参考基因组的项目,获得clean reads后,需要对clean reads进行拼接 以获取后续分析的参考序列。采用Trinity对clean reads进行拼接。
Trinity是一款由 Broad Institute 和 Hebrew University of Jerusalem 合作研发的,针对RNA-seq数据的,高效稳定的转录组拼接软件。其结合了三个独立的 软件模块依次对大量的RNA-seq数据进行了处理拼接,分别为:茧(Inchworm)、 蛹(Chrysalis)、蝶(Butterfly)。主要过程如下:
茧(Inchworm):首先,读入所有的reads的fq文件,然后将fq的reads文件转换成fa 格式的reads文件,并将3'端的reads和5'端的reads进行合并得到both.fa。然后, 将reads按照k-mers (k bp长度的短片段)进行分解,统计kmer的种类和每一类kmer 的数目,根据kmer的频数按照从高到底进行排序。选取频数最高的kmer作为seed开始 向3'端延伸,每次延伸一个碱基,统计延伸之后每种kmer出现的次数,选择kmer频数 最高的那一条kmer作为延伸路径,最后,根据贪婪算法,利用Overlap的关系对k-mers 进行延伸,形成contig序列。
蛹(Chrysalis):聚类所有相似区域大于k-1-mers的 contigs,构成components, 依据不同的components构建de Bruijn graph,并将reads与components进行比对, 进行验证。
蝶(Butterfly):列举转录本 (butterfly),拆分graph 为线性序列,使用reads以及 pairs关系消除错误序列,化简每个component的de Bruijn graph,输出可变剪切亚型 的全长转录本,并梳理对应于旁系同源基因的转录本,最终得到拼接结果文件: TRINITY.fasta。
拼接原理图形展示如下:
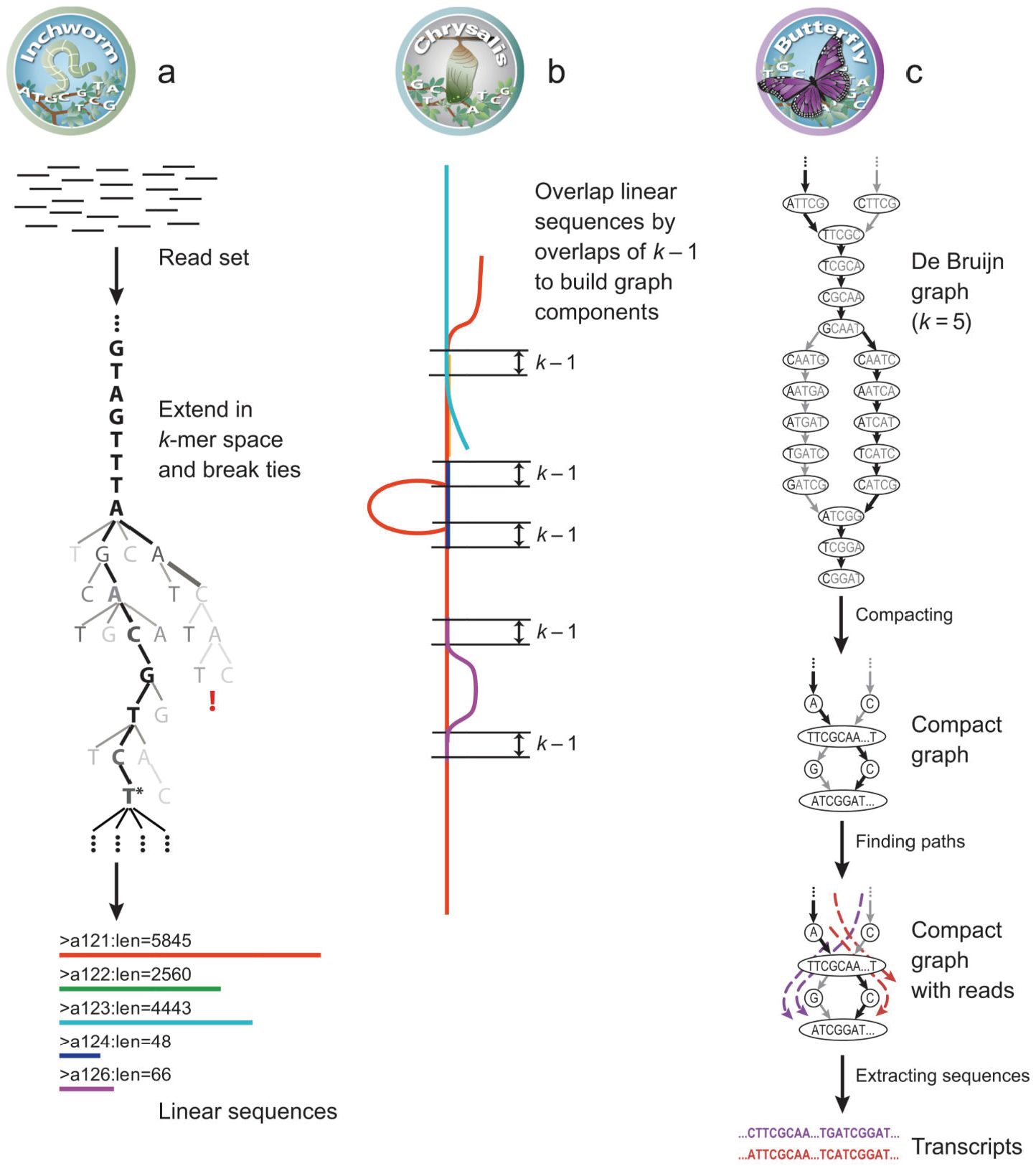
图3-6 Trinity拼接原理图
拼接得到的转录本序列信息以fasta格式储存,见结果文件/customer_files/ 03TranscriptomeAssembly/03.01AssembledTranscriptome/ Trinity.fasta。 下图为该文件中的一条序列作为示例:

图3-7 Trinity拼接序列示例
其中大于号>后紧跟转录本的id号,len=后面为转录本的长度,即该转录本的碱基数, path为从 de Bruijn Graph subComponent中经历的路径。其后为该转 录本的碱基序列。每个转录本的id号构成都为c_g_i, 其中c为拼接过程形成的 de Bruijn Graph Component, g为subcomponet,可以看作为广泛意义上 的gene,i 代表转录本isoform。即:c_g为基因ID,c_g_i为c_g基因不同 的转录本。详细解释见Trinity官方网站 [http://trinityrnaseq.github.io]。
3.2.2 Corset层次聚类
Corset是在Trinity拼接基础上,根据转录本间Shared Reads将转录本聚合为许多 cluster,再结合不同样本间的转录本表达水平及H-Cluster算法,将样本间有表 达差异的转录本从原cluster分离,建立新的cluster,最终每个cluster被定义 为“Gene”。该方法可以聚合冗余转录本,并提高差异表达基因的检出率, Corset官方网站https://code.google.com/p/corset-project/。其中在每 个聚类中选取出最长的转录本作为该聚类中的代表序列,定义为unigene序列。
聚类后的结果文件和选取的unigene序列文件均以fasta格式储存,分别见结果文件 /customer_files/03TranscriptomeAssembly/03.02Corset/ clusterSeq.fa和/customer_files/03TranscriptomeAssembly/03.02Corset/ unigeneSeq.fa。 下图为unigeneSeq.fa文件中的一条序列作为示例:

图3-8 Unigene序列示例
其中大于号>后紧跟序列id号,Clusters-X.Y,X为super_cluster ID:转录本有相同 reads比对上的会有相同的super_cluster ID,Y为tsuper_cluster ID 中的cluster number。
3.2.3 拼接转录本长度分布
将 Trinity 拼接得到的转录本序列,作为后续分析的参考序列。以Corset层次聚类后 得到最长Cluster序列进行后续的分析。对转录本及unigene序列长度分别进行统计, 见结果文件 customer_files/03TranscriptomeAssembly/03.03AssemblyINFO/。
拼接转录本和unigene长度分布情况如下图所示:
图3-9 转录本和unigene在每个区间内序列个数
注:
Length Interval:横坐标为长度区间;
count:纵坐标为对应长度区间内的转录本或unigene的数量。
3.2.4 转录本质量评估
BUSCO[4](Benchmarking Universal Single-Copy Orthologs)评估是使用单拷贝 直系同源基因库,结合tblastn、augustus和hmmer等软件对组装得到的转录本进行评估, 以此评估转录本组装的完整性。我们采用BUSCO软件对拼接得到的Trinity.fasta, unigene.fa和cluster.fasta进行拼接质量的评估,根据比对上的比例、完整性, 来评价拼接结果的准确性和完整性,有关BUSCO的介绍参见下面链接: http://busco.ezlab.org/。这个结果是在文件夹 customer_files/03TranscriptomeAssembly/03.04AssemblyAssesment/中。
其评估结果可视化如下图所示:

图3-10 BUSCO拼接完整性评估
注:
S: Complete Single-Copy BUSCOs
D: Complete Duplicated BUSCOs
F: Fragmented BUSCOs
M: Missing BUSCOs
n: Total BUSCO groups searched
3.3 常用功能数据库注释
3.3.1 功能注释数据库介绍
为了获得gene的功能信息,需要对unigene序列进行功能注释。为使注释信息全面可靠, 这里对unigene序列进行了七大数据库的基因功能注释,包括:Nr,Nt,KOG, Swiss-prot,Uniprot,KEGG,GO。
七大数据库简介
| 数据库 | 简介 | 详细信息 |
|---|---|---|
| Nr | NCBI non-redundant protein sequences,NCBI官方的蛋白序列数据库 | 包括了GenBank基因的蛋白编码序列,PDB(Protein DataBank)蛋白数据库、SwissProt蛋白序列及来自PIR(Protein Information esource)和PRF(Protein Research Foundation)等数据库的蛋白序列。 |
| Nt | NCBI nucleotide sequences,NCBI官方的核酸序列数据库 | 包括了GenBank,EMBL和DDBJ(但不包括EST,STS,GSS,WGS,TSA,PAT,HTG序列)的核酸序列。 |
| Uniprot | Universal Protein是包含蛋白质序列,功能信息,研究论文索引的蛋白质数据库 | Uniprot(Universal Protein)是包含蛋白质序列,功能信息,研究论文索引的蛋白质数据库,整合了包括EBI( European Bioinformatics Institute),SIB(the Swiss Institute of Bioinformatics),PIR(Protein Information Resource)三大数据库的资源。 |
| KOG/COG | COG: Clusters of Orthologous Groups of proteins KOG: euKaryotic Ortholog Groups | KOG和COG都是NCBI的基于基因直系同源关系,其中COG针对原核生物,KOG针对真核生物。COG/KOG结合进化关系将来自不同物种的同源基因分为不同的Ortholog簇,目前COG有4873个分类,KOG有4852个分类。来自同一ortholog的基因具有相同的功能,这样就可以将功能注释直接继承给同一COG/KOG簇的其他成员。详见http://www.ncbi.nlm.nih.gov/COG/。 |
| Swiss-Prot | A manually annotated and reviewed protein sequence database | 搜集了经过有经验的生物学家整理及研究的蛋白序列。详见http://www.ebi.ac.uk/uniprot/。 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes | 系统分析基因产物和化合物在细胞中的代谢途径以及这些基因产物的功能的数据库。它整合了基因组、化学分子和生化系统等方面的数据,包括代谢通路(KEGG PATHWAY)、药物(KEGG DRUG)、疾病(KEGG DISEASE)、功能模型(KEGG MODULE)、基因序列(KEGG GENES)及基因组(KEGG GENOME)等等。KO(KEGG ORTHOLOG)系统将各个KEGG注释系统联系在一起,KEGG已建立了一套完整KO注释的系统,可完成新测序物种的基因组或转录组的功能注释。详见http://www.genome.jp/kegg/ 。 |
| GO | Gene Ontology,一套国际标准化的基因功能描述的分类系统 | GO分为三大类ontology:生物过程(Biological Process)、分子功能(Molecular Function)和细胞组分(Cellular Component),分别用来描述基因编码的产物所参与的生物过程、所具有的分子功能及所处的细胞环境。GO的基本单元是term,每个term有一个唯一的标示符(由“GO:”加上7个数字组成,例如GO:0072669);每类ontology的term通过它们之间的联系(is_a,part_of,regulate)构成一个有向无环的拓扑结构。详见http://www.geneontology.o |
为了方便了解项目的注释率情况,在分析中对本项目数据在七大数据库中的注释成功率进行 统计,见文件/customer_files/04FunctionAnnotation/ 04.01AnnotationResult/annoCount.txt。具体每个数据库注释结果见文件 customer_files/04FunctionAnnotation/04.01AnnotationResult/ 。
在7大数据库注释结果中选出5个数据库绘制的venn图,结果如下:

图3-11 常用功能注释数据库注释结果韦恩图
注:不同颜色表示不同数据库
3.3.2 Nr注释结果
通过与Nr库进行比对注释,可以获取本物种基因序列与近缘物种基因序列的相似性以及本物 种基因的功能信息。NR数据库注释结果完整表格见结果文件 /customer_files/04FunctionAnnotation/04.01AnnotationResult/ NRAnnoInfo.txt 。
本项目对NR数据库比对结果进行统计,获得unigene在各个物种的结果,进而对unigene注释物种进行排序和统计,并绘制比对结果最多的五个物种,并把其余物种比对结果合并到一起,定义为others。统计结果在以下饼图中进行展示:

图3-12 NR数据库比对物种统计
本项目对NR数据库比对结果进行统计,分别绘制evalue分布饼图和序列相似度分布饼图。

图3-13 evalue分布

图3-14 序列相似度分布
3.3.3 GO 注释
对基因进行GO注释后,将注释成功的基因按照GO的三个大类(BP Biological process, CC Cellular component, MF Molecular Function)的下一层级进行分类,如下 图所示,原图片见结果文件 customer_files/04FunctionAnnotation/04.02Goclassification/ 。

图3-15 GO注释分类统计
注:
图中横坐标为GO Term,纵坐标为注释到该Term的基因数目; 因注释到的GO Term较多,每个类别选取前20个作为示例
3.3.4 KOG注释
KOG 分为 25个 group, 将 KOG 注释成功的基因按 KOG 的 group 进行分类, 如下图所示,原图片见结果customer_files/04FunctionAnnotation/04.03KOGclassification/ 。

图3-16 KOG 注释分类统计
注:
A RNA processing and modification
B Chromatin structure and dynamics
C Energy production and conversion
D Cell cycle control, cell division, chromosome partitioning
E Amino acid transport and metabolism
F Nucleotide transport and metabolism
G Carbohydrate transport and metabolism
H Coenzyme transport and metabolism
I Lipid transport and metabolism
J Translation, ribosomal structure and biogenesis
K Transcription
L Replication, recombination and repair
M Cell wall/membrane/envelope biogenesis
N Cell motility
O Posttranslational modification, protein turnover, chaperones
P Inorganic ion transport and metabolism
Q Secondary metabolites biosynthesis, transport and catabolism
R General function prediction only
S Function unknown
T Signal transduction mechanisms
U Intracellular trafficking, secretion, and vesicular transport
V Defense mechanisms
W Extracellular structures
Y Nuclear structure
Z Cytoskeleton
横坐标为KOG的分类,纵坐标count(%)为该分类内注释到的基因占所有注释到的基因的百分比
3.3.5 KEGG注释
对基因做KO注释后,可根据它们参与的KEGG代谢通路进行分类,如下图所示, 原图片见结果文件 customer_files/04FunctionAnnotation/04.04KEGGclassification/ 。

图3-17 KEGG 代谢通路分类统计
注:纵坐标为注释到的KEGG通路,横坐标为注释到通路的基因比例
在此进行功能注释汇总,在表格中对同一基因的不同数据库结果进行汇总,汇总包括geneID、 gene length、NR ID、NR Score、 NR Evalue、NR Description、NT ID、NT Score、NT Evalue、NT Description、 KO ID、KO Name、Swissprot ID、Swissprot Score、Swissprot Evalue、 Swissprot Description、Gene Ontology Biological Pathway、 BP Description、Gene Ontology Cellular Component、 CC Description、Gene Ontology Molecular Function、MF Description、 KOG ID、KOG Description等信息。该表格列数较多,不对其进行展示,表格文件在文件夹 customer_files/04FunctionAnnotation/中。
3.4 CDS预测
CDS是编码一段蛋白产物的序列。预测基因CDS后,根据需要将核酸序列翻译成氨基酸序列, 从而可以用软件预测它的蛋白质结构,推测它的功能或者蛋白类型,另外还可以用来辅助 设计qPCR引物或构建蛋白序列库。
CDS预测有两种常见的方法:一是基于同源序列预测,选取阅读框;二是基于一定的模型预测。
3.4.1 CDS预测
CDS预测分为两步:1.将Gene按照NR蛋白质库,Swissprot蛋白库的优先顺序进行比对, 若比对上,则根据比对结果从蛋白库中提取出蛋白序列;2.对于没有比对上NR蛋白库和Swissprot蛋白库的序列,则使用OrfPredictor软件预测其ORF,从而获得这部分编码的核酸序列和氨基酸序列。详细文件见结果文件customer_files/05CDSprediction/05.01CDSprediction/ 。
比对到NR或Swissprot蛋白库的基因CDS结果示例: 07D_matchSeqCDS.fasta.
比对到NR或Swissprot蛋白库的基因氨基酸结果示例: 07E_matchSeq.pep.fasta.
预测的基因CDS序列示例: unigeneNotMatch.orf.fasta.
预测的基因氨基酸序列示例: unigeneNotMatch.nr.orf.pep.
注,在文件unigeneNotMatch.nr.orf.pep中:
Cluster-X.X:基因ID
+3:预测的6个ORF分别以-3,-2,-1,+1,+2,+3表示,此处指明对应的ORF
834和956:该氨基酸序列对应的CDS在该基因核酸序列中的起始终止位置
3.4.2 基因转录因子分析
植物转录因子预测使用iTAK软件,其基本原理是,利用database中分类定义好的 TF(transcription factor) family及规则,通过hmmscan鉴定TF。
动物转录因子鉴定使用动物转录因子数据库——animalTFDB 2.0,根据转录因子家 族的pfam文件,利用hmmsearch进行鉴定。基因转录因子预测结果见文件 customer_files/05CDSprediction/05.02TFprediction/。 其中转录因子分类信息见文件 tf_classification.txt。
注,在文件tf_classification.txt中:
geneID:预测为转录因子对应的geneID
Subfamily:植物转录因子特有的子家族分类(动物无此列信息)
TF/TR:转录因子或转录调控子
family:转录因子家族信息
若项目中的物种为动物,则预测的转录因子文件为: unigeneSeq.TF.txt。
3.5 基因表达相关分析
3.5.1 基因表达水平分析
3.5.1.1 参考序列比对
将Trinity拼接得到的Unigene序列作为参考序列(Ref),将每个样品的clean reads 往Ref上进行比对,分别过滤掉比对质量低于10的reads,非成对比对的reads,比对到 基因组多个区域的reads。比对过程我们采用了RSEM[7]软件,RSEM中使用bowtie2的 默认参数。比对统计结果见文件customer_files/06GeneExprQuantification/ 06.01.GeneExprQuantification/ RSEM_count.txt。
注,在文件RSEM_count.txt中:
sample name:样本名称
total_reads:样本总reads数量
total_map:能比对到Ref的reads数量
3.5.1.2 基因表达水平统计表
RSEM对bowtie的比对结果进行统计,比对到某基因的reads数目称为read count, 在这里RSEM并不是直接统计比对上的reads数目,而是引入expected_count的概念 对这一数目进行表示。在RSEM结果中,expected_count指来自同一基因或转录本的 reads的后验概率的和。我们对expected_count结果进行提取合并,获得各个样本的 expected_count值,结果见文件 customer_files/06GeneExprQuantification/06.01.GeneExprQuantification/ ExceptCount.txt。
在RNA-seq技术中,TPM(Transcripts Per Kilobase of exon model per Million mapped reads) 是每千个碱基的转录每百万映射读取的Transcripts,是目前流程的基因表达水平估算方法, 同时RSEM软件计算了各个样本的TPM值,我们对其进行提取合并,结果见文件 customer_files/06GeneExprQuantification/06.01.GeneExprQuantification/ TPM.txt。
3.5.1.3 基因TPM密度分布图
TPM密度分布整体体现了各样品的基因表达模式。图形呈非标准正态分布,区域面积均为1, 代表概率的总和为1,密度分布曲线的峰值代表该表达量水平的基因数最多。 原图见结果文件 customer_files/06GeneExprQuantification/06.01.GeneExprQuantification/ , 下图为其中一个样本,作为示例。

图3-18 样本基因表达TPM密度分布图
注:
log10(TPM):横坐标为TPM值的对数值;
density:横坐标对应数值的密度。
3.5.1.4 不同实验条件下,基因表达水平对比图
对不同实验条件下基因的TPM做密度分布图及厢式图,进而能在整体水平上检查不同实验条件 下TPM分布的情况。原图见结果文件 customer_files/06GeneExprQuantification/06.02GeneExpContrast/ , 下图作为示例。

图3-19 不同实验条件下的TPM分布
注:
log10(TPM):各样本TPM值的对数值;
density:各样本横坐标对应值的密度。

图3-20 不同实验条件下TPM厢式图
注:
group:各个分组名称;
log10(TPM+1):TPM值+1的对数值。

图3-21 不同实验条件下TPM区间分布
注:
横坐标为各个分组内不同的TPM区间;纵坐标为横坐标对应区间内的基因数量。
3.5.1.5 样品间相关性检查
样品间基因表达水平相关性是检验实验可靠性和样本选择是合理的重要指标。生物学重复是 生物学实验所必须的,设置生物学重复主要有两个用途:一个是证明所涉及的生物学实验 操作不是偶然,而是可重复的;另一个是为了确保后续的差异基因分析得到更可靠的结果。 样品间基因表达水平相关性是检验实验可靠性和样本选择是否合理的重要指标。 相关系数越接近1,表明样品之间表达模式的相似度越高。若样品中有生物学重复, 通常生物重复间相关系数要求较高。Encode计划建议皮尔逊相关系数的平方(R2) 大于0.92(理想的取样和实验条件下)。具体的项目操作中,我们要求生物学重复样品间 R2至少要大于0.8。根据样本所有基因的TPM值计算组内及组间的相关性系数,绘制成热图, 可直观显示组间样本差异及组内样本重复情况。样本相关性热图如下图所示, 原图见结果文件 customer_files/06GeneExprQuantification/06.03sampleCor/ 。

图3-22 样品间相关系数热图
3.5.2 差异分析
在转录组中,确定某个基因在不同的样品中的表达量是否有差异是分析的核心内容之一。 获得基因表达量后,即可对表达数据进行统计学分析,进而筛选不同样本之间显著差异的基因。 差异分析主要分为三个步骤:
首先对原始的read count进行标准化(normalization),主要是对测序深度的校正。 然后通过统计学模型进行假设检验概率(P-value)的计算,最后进行多重假设检验校正 (BH),得到FDR值(错误发现率,padj是其常见形式,以下皆使用padj代表FDR)。
针对不同的实验情况,我们选用合适的软件进行基因表达差异显著性分析,具体如下表所示。
表3.3表达差异分析所用软件及差异基因筛选标准

一般来说,如果一个基因在两组样品中的表达量差异达到两倍以上,我们认为这样的基因是具 有表达差异的。为了判断两个样品之间的表达量差异究竟是由于各种误差导致的还是本质差异, 我们需要对所有基因在这两个样本中的表达量数据进行假设检验。而转录组分析是针对成千上 万个基因进行的,这样会导致假阳性的累积,基因数目越多,假设检验的假阳性累积程度会越高, 所以引入padj对假设检验的P-value进行校正,从而控制假阳性的比例。
3.5.2.1 差异基因列表
我们对每个比较组合进行差异显著性分析,最得到差异表达基因列表,如下表所示。该项目中 含有生物学重复,所以使用DEseq2计算差异表达基因,原文件见结果文件 customer_files/07DiffExprAnalysis/group*vsgroup*.txt。
3.5.2.2 差异基因统计表
每个比较组合的差异基因(包括上调和下调)数目统计以及筛选差异的标准如下表所示, 原文件见结果文件customer_files/07DiffExprAnalysis/ dataUpDown.txt。
对每个比较组合的差异基因数(包含上调和下调)用柱状图进行展示,原文件见结果文件 customer_files/07DiffExprAnalysis/ 。

图3-23 差异比较组合差异基因数目统计柱状图
注:
横坐标为不同对比组的所有基因、下调基因和上调基因;纵坐标为横坐标对应的差异基因的数量。
3.5.2.3 差异基因火山图
火山图可直观显示表达差异显著性基因的整体分布情况,横坐标表示基因在不同样本中的 表达倍数变化(log2FoldChange),纵坐标表示表达差异的显著性水平(- log10padj)。 若比较组合无表达差异显著性基因,默认调整筛选表达差异显著性的阈值进行火山图的绘制。 上调基因用红色点表示,下调基因用蓝色点表示,如下图所示,原文件见结果文件 customer_files/07DiffExprAnalysis/ 。

图3-24 差异基因火山图
注:
横坐标为log2(Fold Change),即fold change值的对数值;纵坐标为-log10(padj),即矫正P值的对数的相反数。
3.5.2.4 差异基因韦恩图
韦恩图可展示不同比较组合间差异基因的重叠情况,通过韦恩图,可筛选某几个比较组合共有 或独有的差异基因。若只有一个差异比较组合,我们默认绘制该比较组合处理组与对照组的共 表达基因韦恩图。韦恩图中圈内所有数字之和代表该比较组合差异基因总数,重叠区域表示组 合间共有的差异基因个数,具体如下图所示,原文件见结果文件 customer_files/07DiffExprAnalysis/。

图3-25 差异基因韦恩图
3.5.2.5 差异基因聚类
将所有比较组的差异基因取并集之后作为差异基因集。两组以上的实验, 可对差异基因集进行聚类分析,将表达模式相近的基因聚在一起。 我们采用主流的层次聚类对基因的TPM值进行聚类分析,对行(row)进行均一化处理 (Z-score)。热图中表达模式相近的基因或样本会被聚集在一起,每个方格中的颜色 反映的不是基因表达值,而是表达数据的行进行均一化处理后得到的数值,所以热图中的 颜色只能横向比较(同一基因在不同样本中的表达情况),而不能纵向比较(同一样本不 同基因的表达情况)。该项目中对不同组间进行聚类,由于差异基因较多会导致热图画图 失败,所以分析时选取 6000/组别数 个基因进行热图绘制,原文件见结果文件 customer_files/07DiffExprAnalysis/ 。

图3-26 差异表达基因聚类热图
3.5.3 富集分析
对于转录组分析而言,我们往往会得到成百上千的差异基因,这无疑增加了后续分析的复杂度。 因此,可以将这些差异基因按照不同的功能类别进行分类,即对差异基因进行富集分析。 通过富集分析,可以找到不同条件下的差异基因与哪些生物学功能或通路显著性相关, 从而揭示和理解生物学过程的基本分子机制。
我们采用diamond和clusterProfiler软件对差异基因集进行GO功能富集分析, KEGG通路富集分析等。富集分析基于超几何分布原理,其中差异基因集为差异显著分析所得 差异基因并注释到GO或KEGG数据库的基因集,背景基因集为所有进⾏差异显著分析的基因并 注释到GO或KEGG数据库的基因集。富集分析结果是对每个差异比较组合的所有差异基因集、 上调差异基因集、下调差异基因集进⾏富集。本报告中展示的表格是选取某⼀个比较组合的 富集分析结果,结果文件中包含所有组合的富集分析结果。

3.5.3.1 GO功能富集
3.5.3.1.1 GO富集列表
GO(Gene Ontology)是描述基因功能的综合性数据库(http://www.geneontology.org/), 可分为生物过程(Biological Process)和细胞组成(Cellular Component) 分子功能(Molecular Function)三个部分。GO 功能显著性富集分析给出与基因组背景相比, 在差异表达基因中显著富集的 GO 功能 条目,从而给出差异表达基因与哪些生物学功能显著相关。 该分析首先把所有差异表达基因向 Gene Ontology 数据库的各个 term 映射,计算每个 term 的基因数目,然后找出与整个基因组背景相比,在差异表达基因中显著富集。GO富集分析方法为 clusterProfiler的enricher函数,该方法使用超几何分布检验的方法获得显著富集的GO Term。 统计被显著富集的各个GOterm中的基因数,以柱状图的形式展示。GO富集以padj小于0.05为显著富集, 富集结果如下表所示。该部分结果文件见文件夹: customer_files/08EnrichAnalysis/08.01Goenrichment/ , 结果文件以go_rich开头。
3.5.3.1.2 GO富集柱状图
根据GO富集分析结果,在GO的三个分类中(cellular_component:细胞组分; biological_process:生物学过程;molecular_function:分子功能) 分别统计每个分类上调基因、下调基因及所有基因的数目,以柱状图展示。 该部分结果图片见文件夹:customer_files/08EnrichAnalysis/08.01Goenrichment。

图3-27 差异基因GO富集柱状图
注:
横坐标为GO数据库GO Term的三个分类;纵坐标为对应分类内的上调基因和下调基因个数。

图3-28 差异表达基因GO Term注释网络图
注:图中节点为差异表达基因注释到的GO Term,节点颜色为富集分析注释的p值,节点大小为注释到该节点的基因个数。节点之间连线表示这两个节点代表的GO Term之间具有共享的基因。

图3-29 差异表达基因与GO Term网络图
注:图中浅黄色节点为差异表达基因注释到的GO Term,灰色节点为差异表达基因。基因和GO Term之间的连线表示该基因注释到该GO Term。

图3-30 差异表达基因聚类热图(环形布局)
注:图中浅黄色节点为差异表达基因注释到的GO Term,灰色节点为差异表达基因。基因和GO Term之间的连线表示该基因注释到该GO Term。节点之间不同颜色连线代表从不同GO Term发出的连线。
3.5.3.1.3 topGO有向无环图分析
topGO有向无环图能直观展示差异基因富集的GO term及其层级关系。有向无环图为差异基因 GO富集分析的结果图形化展示方式,分支代表包含关系,从上至下所定义的功能范围越来越具体。 对GO三大分类(CC细胞成分,MF 分子功能,BP生物学过程)的每一类都取富集程度最高的前 10位作为有向无环图的主节点,用方框表示,并通过包含关系将相关联的GO Term一起展示, 颜色的深浅代表富集程度,颜色越深代表富集程度越高。该部分结果文件见文件夹 customer_files/08EnrichAnalysis/08.02DAG/。

图3-31 差异基因topGO
注:每个圆圈代表一个GO Term,放大方框中内容从上到下代表的含义依次是: GO term的id、GO的描述、GO富集的矫正P值、该GO下差异基因数目
3.5.3.2 KEGG通路富集
3.5.3.2.1 KEGG通路富集列表
在生物体内,不同基因相互协调行使其生物学功能,通过Pathway显著性富集能确定差异 表达基因参与的最主要生化代谢途径和信号转导途径。
KEGG(Kyoto Encyclopedia of Genes and Genomes)是有关Pathway的主 要公共数据库,其中整合了基因组化学和系统功能信息等众多内容。信息分析时, 我们将KEGG数据库按照动物,植物,真菌等进行分类,依据研究物种的种属选择相应的类别进行分析。 Pathway显著性富集分析以KEGG Pathway为单位,应用超几何检验,找出差异基因相对 于所有有注释的基因显著富集的pathway,KEGG通路富集同样以padj小于0.05作为显著 性富集的阈值。该部分全部结果存在文件夹 customer_files/08EnrichAnalysis/08.03KEGGenrichment中, 结果文件以kegg_tmp开头。
3.5.3.2.2 KEGG通路富集气泡图
差异基因KEGG富集气泡图是KEGG富集分析结果的图形化展示方式。在此图中, KEGG富集程度通过GeneRatio、p.adjust和富集到此通路上的基因个数来衡量。 其中GeneRatio指差异表达的基因中位于该pathway条目的基因数目与所有有注释 基因中位于该pathway条目的基因总数的比值。p.adjust是做过多重假设检验校正之后的 Pvalue,p.adjust的取值范围为[0,1],越接近于零,表示富集越显著。 我们挑选了富集最显著的20条pathway条目在该图中进行展示, 若富集的pathway条目不足20条,则全部展示。该部分的结果见文件夹 customer_files/08EnrichAnalysis/08.03KEGGenrichment/。

图3-32 KEGG通路富集气泡图
注:KEGG pathway富集气泡图:纵轴表示pathway名称,横轴表示pathway对应的GeneRatio,p.adjust的大小用点的颜色来表示,p.adjust越小则颜色越接近红色,每个pathway下包含的差异基因的多少用点的大小来表示。
3.5.3.2.3 富集KEGG通路图
将差异基因富集出的通路图展示出来,示意如下。为了便于查看差异基因在通路图中的分布情况,我们将差异基因标注到通路图中,分别用红色和绿色表示该KO节点含有上调基因和下调基因。该部分结果见文件夹customer_files/08EnrichAnalysis/08.03KEGGenrichment。
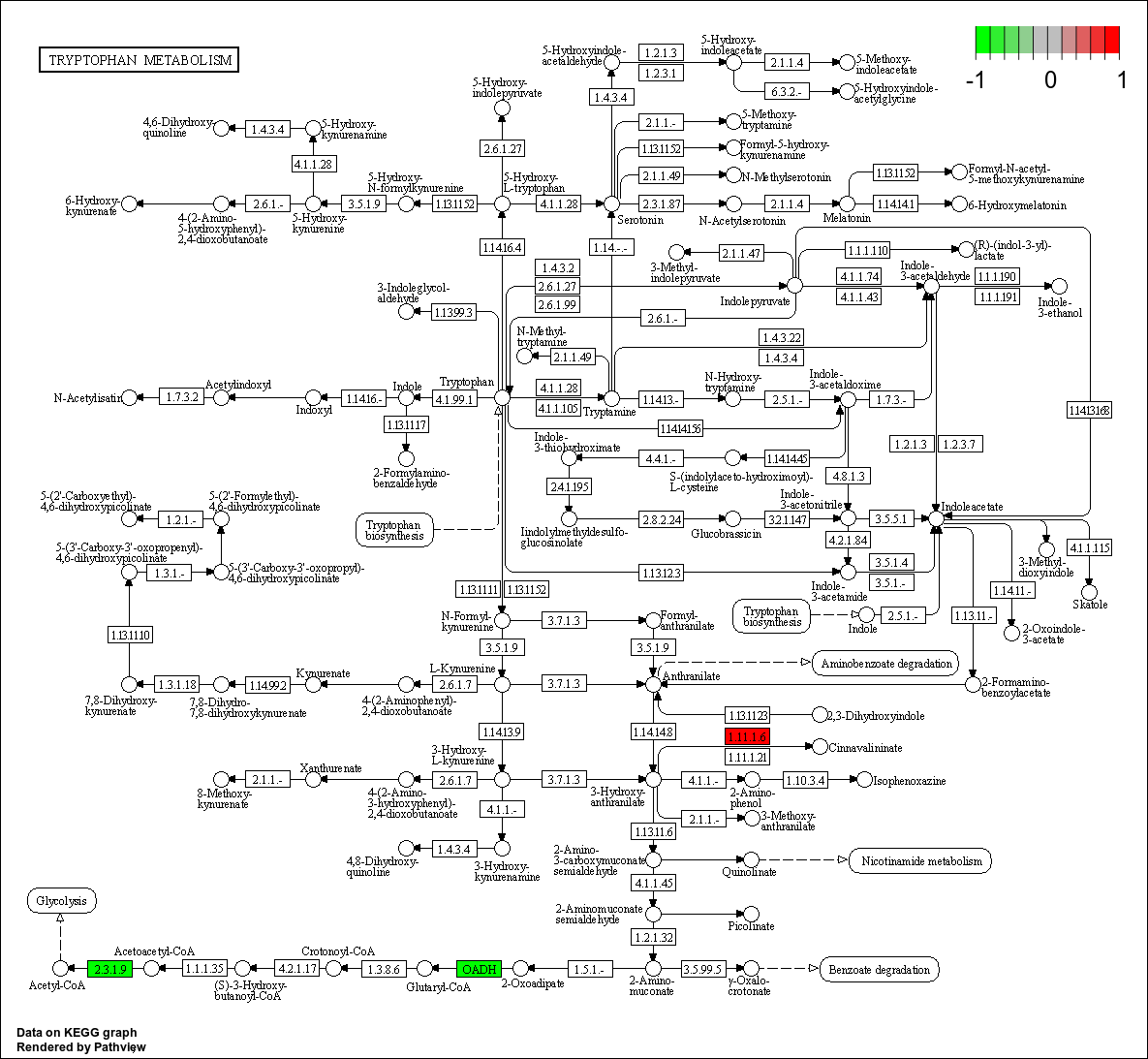
图3-33 KEGG富集分析通路图
这里对差异表达基因及其功能注释部分进行汇总,汇总信息包括gene_id、groups、 log2FoldChange、padj及功能注释汇总部分,表格列数较多,不进行展示。 表格数据在文件夹 customer_files/08EnrichAnalysis/中,表格文件以group开头。
3.5.4 基因共表达网络分析
WGCNA(weighted geneco-expression network analysis)算法是一种构建基因共表达网络的典型系统生物学算法,该算法基于高通量的基因信使RNA(mRNA)表达数据,被广泛应用于国际生物医学领域。WGCNA算法首先假定基因网络服从无尺度分布,并定义基因共表达相关矩阵、基因网络形成的邻接函数,然后计算不同节点的相异系数,并据此构建分层聚类树(hierarchical clustertingtree)。该聚类树的不同分支代表不同的基因模块(module),模块内基因共表达程度高,而分数不同模块 的基因共表达程度低。最后,探索模块与特定表型或疾病的关联关系,以此达到鉴定目标的靶点基因、基因网络的目的。基因共表达网络分析的结果位于文件夹customer_files/08EnrichAnalysis中。
3.5.4.1 软阈值选择
WGCNA首先会计算任意两个基因之间的相关系数(Person Coefficient)。为了衡量两个基因是否具有相似表达模式,一般需要设置阈值来筛选,高于阈值的则认为是相似的,但如果将阈值设为0.8,那么很难说明0.8和0.79两个是有显著差别的。因此,WGCNA分析时采用相关系数加权值,即对基因相关系数取N次幂,这种处理方式强化了强相关,弱化了弱相关或负相关,使得网络中的基因之间的连接服从无尺度网络分布(scale-freenetworks),更具有生物意义。下图中图形的横轴均代表权重参数β,也就是软阈值。纵轴代表对应的网络中相关系数的平方。相关系数的平方越高,说明该网络越逼近无尺度网络的分布,我们设定相关系数的平方的阈值是0.8。最佳的β值就是后续分析使用的软阈值。

图3-34 软阈值选择示意图
3.5.4.2 模块层次聚类
WGCNA分析会根据基因间表达量的相关性构建聚类树,并划分模块。图中每一种颜色表示一种颜色对应聚类树上的每个基因属于同一个模块,如果某些基因在一个生理过程或不同组织中总是具有相类似的表达变化,那么这些基因在功能上可能相关,可以把他们定义为一个模块(module)对于上半部分的树图,纵向距离代表两个节点间(基因间) 的距离,横向距离无意义。

图3-35 模块层次聚类树图
3.5.4.3 模块间相关性热图
模块间相关性热图可分为两部分,上部分根据模块特征值(eigengene)对模块进行聚类。纵坐标代表节点的相异程度;下半部分图形中每一行和列代表一个模块。方块颜色越深(越红),相关性越强;方块颜色越浅,相关性越弱。

图3-36 模块间相关性热图
3.5.4.4 样品和模块间相关性热图
一般而言,如果某模块与样品的相关性显著高于其它模块,说明这一个模块可能与该样品存在最强的关联关系,如下图所示。

图3-37 样品和模块间相关性热图
注:横坐标为样本,纵坐标为模块,每个格子的数字代表模块与样品的相关性,该数值越接近1,表示模块与样品正相关性越强;越接近-1,表示模块与样品负相关性越强。括号里的数字代表显著性Pvalue,该数值越小,表示显著性越强。
3.5.4.5 模块基因表达模式
模块基因表达模式图可分为两部分查看,表头注释为模块名,上图左侧为基因名,上图横坐标为样本名,上图为模块中基因在不同样本中的表达量热图,红色为高表达,绿色为低表达,通过上图可直观看出模块中的基因在不同样本中的表达趋势。下图为模块特征值在不同样本中的表达模式,横坐标为样本名,通过下图柱状图的展示,可直观看出哪个样本中的gene在该模块下普遍高表达。如下图可知:

图3-38 模块基因表达模式
3.5.4.6 基因连通性
一个基因与其他基因的连接程度(通常只在模块内计算)。常称为connectivity或degree,或用数字k表示。一般而言,在一个模块中,连通性(k值)排名 靠前的基因可认为核心基因(hub gene)。k值排名靠前本身已经表明它们处于中心枢纽的位置。连通性的结果如下表所示:

3.5.5 差异基因蛋白互作网络分析
3.5.5.1 差异基因蛋白互作网络分析
我们主要应用STRING蛋白质互作数据库(http://string-db.org/) 中的互作关系,针对数据库中包含的物种,直接从数据库中提取出目标基因集(比如差异基因list)的互作关系构建网络;针对数据库中不包含的物种,我们首先将目标基因集序列应用blastx[6](evalue设为1e-10)比对到string数据库中包含的参考物种蛋白质序列上,并利用参考物种的蛋白质互作关系构建互作网络。
我们提供差异基因蛋白互作网络数据文件,此文件可以直接导入Cytoscape软件进行可视化编辑。Cytoscape软件使用方法可参考我们提供的使用说明文档(CytoscapeQuickStart.pdf)。客户可以针对一些网络的拓扑属性进行统计和标示作图,比如:互作网络图中节点(node)的大小与此节点的度(degree)成正比,即与此节点相连的边越多,它的度越大,节点也就越大,这些节点在网络中可能处于较为核心的位置。节点的颜色与此节点的聚集系数(clustering coefficient)相关,颜色梯度由绿到红对应聚集系数的值由低到高;聚集系数表示此节点的邻接点之间的连通性好坏,聚集系数值越高表示此节点的邻接点之间的连通性越好等等。根据不同的研究目的和需求,客户还可以在网络图中进行调整节点位置和颜色、标注表达量水平等操作。需要注意的是,通过blast比对得到的结果不能保证较好的准确性,这部分的工作只是给客户提供参考,辅助客户发现一些可能的重要的基因。按我们提供的使用说明将文件导入Cytoscape软件后的效果图如下。根据差异表达基因提取的互作网络见结果文件customer_files/09StringNet/。

图3-39 差异基因蛋白网络互作图
注:图中每个节点表示一个蛋白,每一个连线表示蛋白的互作
3.6 SNP和InDel分析
3.6.1 SNP和InDel分析
SNP全称Single Nucleotide Polymorphisms,是指在基因组上由单个核苷酸变异形成的遗传标记,其数量很多,多态性丰富。从理论上来看每一个SNP 位点都可以有4 种不同的变异形式,但实际上发生的只有两种,即转换和颠换,二者之比为1:2。SNP在CG序列上出现最为频繁,而且多是C转换为T,原因是CG中的C常为甲基化的,自发地脱氨后即成为胸腺嘧啶。一般而言,SNP是指变异频率大于1%的单核苷酸变异。InDel(insertion-deletion)是指相对于参考基因组,样本中发生的小片段的插入缺失,该插入缺失可能含一个或多个碱基。
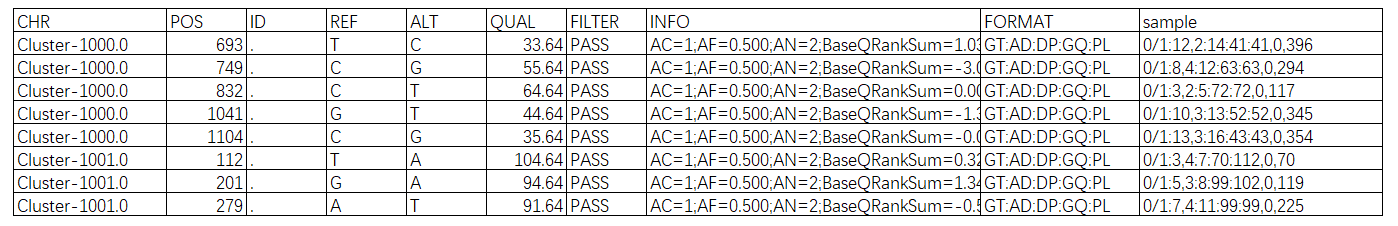
我们使用samtools对比对结果进行染色体坐标排序,使用变异检测软件GATK4对比对结果进行重复标记,然后分别进行SNP Calling和InDel Calling,并对原始结果进行过滤,得到如下表形式的分析结果。其中InDel分析结果每列的含义和SNP结果是一致的。所有结果见文件夹: customer_files/10SNPIndelAnalysis/,各个样本的结果文件在对应的文件夹中单独存放。示例:bowtie2.sorted.add.markdup.snp.filtered.PASS.vcf
表3.4 SNP和Indel结果示例
3.7 SSR分析
3.7.1 SSR分析
简单重复序列(Simple Sequence Repeat,简称SSR), 是指重复单位长度和碱基组成基本一致,只 有少数基因出现微小的差异的一类短串联重复序列。 真核生物基因组中存在大量重复频率极高而顺序复杂性极低的串联重复序列, 这些串联重复序列的突变情况在不同物种、同一物种的不同位点或同一 位点的不同等位基因间存在很大差异。
我们采用MISA软件1.0版,默认参数;对应的各个unit size的最少重复次数分别为: 1-10,2-6,3-5,4-5,5-5,6-5(如:1-10,以单核苷酸为重复单位时, 其重复数至少为10才可被检测到;2-6,以双核甘酸为重复单位时,其最少重复数为6) 对Gene进行SSR检测,详见http://pgrc.ipk-gatersleben.de/misa/misa.html 。 原文件见结果文件 customer_files/11SSRAnalysis/。
我们还对不同SSR类型在基因转录本的密度分布进行统计,结果如下图所示。

图3-40 SSR种类分布
注:A-F指重复次数区间
3.7.2 SSR引物设计
虽然 SSR 在基因组上的位置不尽相同,但是其两端序列多是保守的单拷贝序列, 因此可根据 SSR 两端互补序列来设计引物,通过 PCR 反应扩增出含有 SSR 位点的片段。 由于重复单位的串联重复次数不同,因而能够用 PCR 的方法扩增出不同长度的 PCR 产物, 将得到的产物进行凝胶电泳,即可显示 SSR 位点的 长度多态性。 我们采用 Primer3(2.3.5版,默认参数)进行 SSR 引物设计,并针对预测到的每一个 SSR 位点分别设计了三组引物供客户选用。原文件列数较多,故截断展示,原文件见结果文件 customer_files/11SSRAnalysis/。
四、所用软件的版本

五、 参考文献
1. Trimmomatic: a flexible trimmer for Illumina sequence data. (Trimmomatic)
2. Full-length transcriptome assembly from RNA-Seq data without a reference genome. (Trinity)
3. Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. (Corset)
4. BUSCO: Assessing Genome Assembly and Annotation Completeness. (BUSCO)
5. Fast and sensitive protein alignment using DIAMOND. (DIAMOND)
6. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. (BLAST)
7. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. (RSEM)