温馨提示:请使用火狐或者Chrome的网页浏览器来查看报告
微科盟真核无参转录组云流程结题报告
一 概述
核糖核酸(Ribonucleic acids,RNA)是细胞和生物体内一类重要的生物分子。它们代表基因组表达或输出的情况。RNA种类繁多,每一种RNA都在基因表达中起着重要的作用,并最终将基因型与表型联系起来。信使RNA
( Messenger RNAs, mRNA )是一类重要的RNA,其编码的信息能够用于翻译蛋白质[1]。随着Illumina为代表的二代测序(next generation sequencing ,
NGS)技术逐渐成熟和成本逐渐降低,我们可以越来越方便的通过差异表达分析(differential gene expression, DGE)确定不同实验组之间基因表达水平的定量变化[2]。
确定mRNA表达水平的方法主要分为两类:基于比对的方法(reference-guided approach)和基于组装(De novo transcriptome
assembly)的方法[3]。基于比对的方法首先将所有测序仪产生的测序片段(reads)比对到一个参考基因组上,确定reads来源于哪个基因;而基于组装的方法直接将reads组装为转录本(transcriptome),无需提供参考基因组。当存在缺少参考基因组,测序样本与参考基因组差异过大导致比对率低,参考基因组高度碎片化等无法使用基于比对的方法时,可以考虑使用基于组装的策略。
二 项目流程
无参转录组云流程包含了以下数据分析方法。

三 数据分析结果
FASTQ 文件是一种广泛用于存储生物测序数据(如 DNA、RNA 或蛋白质序列)及其对应质量分数的文本格式。一个典型的fastq示例如下:
@SEQ_ID:1:FC1:1:1:1234:5678
每个测序读数(read)在文件中由 4 行组成,结构如下:
AGCTTGCAGTAGCTAGCTACGATCGATCGATCG
+
!''*((((***+))%%%++)(%%%%).1***-+*''))
- 第一行用于描述read的来源,以 @ 开头,后跟唯一标识符(如测序仪器名称、位置等信息);
- 第二行记录测序得到的实际序列,由核苷酸序列(如 A, T, C, G, N)组成;
- 第三行以 + 开头,分隔第二行和第四行。可选择性包含与头行相同的标识符(通常省略);
- 第四行表示每个碱基的测序质量分数,由 ASCII 字符组成,每个字符对应一个测序分数。
3.1 原始数据质量控制
3.1.1 fastp
为了检测和处理原始的fastq格式测序数据中可能存在的接头、删除过滤低质量片段以及进行基于重叠的reads校正,我们首先使用fastq[5]软件处理原始的测序文件,以保证下游分析输的准确性。在该步骤中进行如下工作:
- 过滤掉低质量,太短和太多N的reads;
- 从头部(5')或尾部(3')计算滑动窗内的碱基质量均值,并切除低质量碱基;
- 删除掉上下机序列可能不稳定导致的头尾低质量碱基;
- 自动识别接头序列并去除接头;
- 对不匹配的碱基对进行矫正;
- 修剪尾部(3')的polyG。
| total_reads | total_bases | q20_bases | q30_bases | q20_rate | q30_rate | read1_mean_length | read2_mean_length | gc_content | |
|---|---|---|---|---|---|---|---|---|---|
| test1 | 57454204 | 8618130600 | 8428027413 | 8137689455 | 0.977941 | 0.944252 | 150 | 150 | 0.594988 |
| test2 | 54911764 | 8236764600 | 8056736993 | 7781646164 | 0.978143 | 0.944745 | 150 | 150 | 0.594646 |
| test3 | 64125598 | 9618839700 | 9412340948 | 9079550743 | 0.978532 | 0.943934 | 150 | 150 | 0.594223 |
| test4 | 63175692 | 9476353800 | 9278603025 | 8973498295 | 0.979132 | 0.946936 | 150 | 150 | 0.594706 |
| test5 | 50016786 | 7502517900 | 7341405253 | 7092158820 | 0.978526 | 0.945304 | 150 | 150 | 0.593369 |
| test6 | 53223146 | 7983471900 | 7800625280 | 7524216923 | 0.977097 | 0.942474 | 150 | 150 | 0.594093 |
| test7 | 58240106 | 8736015900 | 8543486088 | 8251437851 | 0.977961 | 0.944531 | 150 | 150 | 0.594112 |
| test8 | 49113206 | 7366980900 | 7204393625 | 6959591769 | 0.977930 | 0.944701 | 150 | 150 | 0.590951 |
| test9 | 48774614 | 7316192100 | 7162443068 | 6923654964 | 0.978985 | 0.946347 | 150 | 150 | 0.594027 |
| total_reads | total_bases | q20_bases | q30_bases | q20_rate | q30_rate | read1_mean_length | read2_mean_length | gc_content | |
|---|---|---|---|---|---|---|---|---|---|
| test1 | 56506940 | 8362330912 | 8239700960 | 8000981916 | 0.985335 | 0.956788 | 147 | 147 | 0.595270 |
| test2 | 54028798 | 7988164500 | 7872191587 | 7646209050 | 0.985482 | 0.957192 | 147 | 147 | 0.595111 |
| test3 | 63119294 | 9384014016 | 9242165194 | 8959316677 | 0.984884 | 0.954742 | 148 | 148 | 0.594223 |
| test4 | 62259666 | 9214159050 | 9083581445 | 8829271615 | 0.985829 | 0.958229 | 147 | 147 | 0.595230 |
| test5 | 49201674 | 7304485214 | 7197989071 | 6990573683 | 0.985420 | 0.957025 | 148 | 148 | 0.593599 |
| test6 | 52304514 | 7738610262 | 7620283683 | 7392064058 | 0.984710 | 0.955219 | 147 | 147 | 0.594643 |
| test7 | 57308184 | 8481204438 | 8356627507 | 8115799751 | 0.985311 | 0.956916 | 147 | 147 | 0.594598 |
| test8 | 48313112 | 7164572444 | 7058318988 | 6855285195 | 0.985170 | 0.956831 | 148 | 148 | 0.591203 |
| test9 | 48048710 | 7129715694 | 7026608023 | 6824904108 | 0.985538 | 0.957248 | 148 | 148 | 0.594339 |
注:total reads:测序reads总数,
total bases:测序得到的碱基总数,
q20 bases:错误率低于1/10^20的碱基数量,
q20 rate:错误率低于1/10^20的碱基占全部碱基的比例,
q30 bases:错误率低于1/10^30的碱基数量,
q30 rate:错误率低于1/10^30的碱基占全部碱基的比例,
read1 mean length:一端reads的平均长度,
read2 mean length:另一端reads的平均长度,
gc_content:GC碱基的比例。
3.1.2 fastq_screen
样本在存储、运输及测序的过程中均有可能被污染,从而导致测序数据中出现其他物种的转录本(外源污染)或其他类型的RNA序列(内源污染)。我们使用FastQ
Screen[6]检测经过fastp过滤后的序列是否存在上述两类污染。未经污染的情况下,目标物种比对率应占绝对主导(如人类样本>90%比对到人类基因组)。若其他物种(如小鼠、大肠杆菌)的比对率显著(如>5%),可能提示污染。(如人类样本中检测到10%的小鼠比对率
→ 可能实验室交叉污染(如小鼠细胞混入)
fastq_screen的汇总结果如下表所示:
| Human | Mouse | Rat | Drosophila | Worm | Yeast | Arabidopsis | Ecoli | rRNA | MT | PhiX | Lambda | Vectors | Adapters | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| test1_1 | 1.03 | 1.27 | 1.35 | 0.95 | 0.33 | 0.41 | 0.35 | 0.00 | 0.14 | 0.00 | 0.0 | 0.0 | 0.05 | 0.02 |
| test2_1 | 0.91 | 1.09 | 1.19 | 0.83 | 0.26 | 0.32 | 0.29 | 0.00 | 0.10 | 0.01 | 0.0 | 0.0 | 0.03 | 0.01 |
| test3_1 | 0.88 | 1.07 | 1.14 | 0.85 | 0.28 | 0.34 | 0.28 | 0.00 | 0.09 | 0.00 | 0.0 | 0.0 | 0.02 | 0.00 |
| test4_1 | 1.02 | 1.27 | 1.36 | 0.95 | 0.34 | 0.47 | 0.45 | 0.00 | 0.23 | 0.00 | 0.0 | 0.0 | 0.07 | 0.01 |
| test5_1 | 0.86 | 1.04 | 1.13 | 0.75 | 0.28 | 0.33 | 0.31 | 0.00 | 0.16 | 0.00 | 0.0 | 0.0 | 0.08 | 0.01 |
| test6_1 | 1.00 | 1.21 | 1.22 | 0.89 | 0.33 | 0.36 | 0.28 | 0.00 | 0.10 | 0.00 | 0.0 | 0.0 | 0.09 | 0.01 |
| test7_1 | 0.96 | 1.16 | 1.22 | 0.87 | 0.30 | 0.34 | 0.23 | 0.00 | 0.07 | 0.01 | 0.0 | 0.0 | 0.05 | 0.00 |
| test8_1 | 0.95 | 1.19 | 1.24 | 0.90 | 0.29 | 0.36 | 0.29 | 0.00 | 0.12 | 0.00 | 0.0 | 0.0 | 0.04 | 0.00 |
| test9_1 | 0.97 | 1.18 | 1.26 | 0.90 | 0.31 | 0.37 | 0.24 | 0.00 | 0.10 | 0.00 | 0.0 | 0.0 | 0.02 | 0.00 |
| test1_2 | 1.21 | 1.47 | 1.55 | 0.87 | 0.59 | 0.38 | 0.36 | 0.00 | 0.17 | 0.00 | 0.0 | 0.0 | 0.33 | 0.01 |
| test2_2 | 1.12 | 1.39 | 1.43 | 0.80 | 0.52 | 0.31 | 0.30 | 0.01 | 0.12 | 0.00 | 0.0 | 0.0 | 0.32 | 0.00 |
| test3_2 | 1.18 | 1.37 | 1.45 | 0.84 | 0.57 | 0.32 | 0.25 | 0.00 | 0.09 | 0.00 | 0.0 | 0.0 | 0.33 | 0.00 |
| test4_2 | 1.17 | 1.40 | 1.51 | 0.92 | 0.53 | 0.44 | 0.43 | 0.00 | 0.22 | 0.00 | 0.0 | 0.0 | 0.26 | 0.00 |
| test5_2 | 1.16 | 1.40 | 1.46 | 0.84 | 0.57 | 0.38 | 0.32 | 0.00 | 0.16 | 0.00 | 0.0 | 0.0 | 0.33 | 0.01 |
| test6_2 | 1.30 | 1.62 | 1.68 | 0.96 | 0.69 | 0.37 | 0.29 | 0.00 | 0.10 | 0.01 | 0.0 | 0.0 | 0.44 | 0.00 |
| test7_2 | 1.15 | 1.43 | 1.47 | 0.85 | 0.57 | 0.32 | 0.23 | 0.00 | 0.08 | 0.00 | 0.0 | 0.0 | 0.34 | 0.00 |
| test8_2 | 1.25 | 1.52 | 1.60 | 0.88 | 0.63 | 0.41 | 0.30 | 0.00 | 0.13 | 0.00 | 0.0 | 0.0 | 0.36 | 0.00 |
| test9_2 | 1.22 | 1.50 | 1.60 | 0.89 | 0.56 | 0.34 | 0.24 | 0.00 | 0.10 | 0.00 | 0.0 | 0.0 | 0.33 | 0.00 |
3.2 转录本的组装
测序数据的组装通常使用德布鲁因图(de Bruijn graph),该方法用于寻找一个从给定字母表中取出的字母的循环序列,对于这个循环序列,每个可能的一定长度的单词恰好一次作为连续字符串出现一次。
在对测序数据进行组装的过程中,我们将长度为k-1(k小于reads的长度)的短序列(k-1 mer)作为图的节点(node),将分别从前向和后向包含两个节点并且长度为k的序列(k
mer)作为边(edge)构建德布鲁因图,然后找到一个恰好访问图的所有边且长度最小的圈(cycle)[7],即为一个可能的组装。
与从头组装基因组不同,在组装转录本的时候我们得到更多不相关联的圈,然后对每个圈进行独立处理,提取全长异构体,并对旁系同源序列(物种内起源于同一个基因的不同基因,其序列具有一定相似性但其功能可能已经发生分化)进行区分。
3.2.1 基于Trinity的转录本的组装
在云流程中我们使用Trinity[8]软件对转录本进行组装,组装过程可以分为三步:
- Inchworm程序读入测序生成的独特的reads,使用基于贪婪k mer的方法进行快速有效的转录本组装,得到重叠群(contig)。对于多个组装结果共享同一个k mer的情况(可变剪切、旁系同源、等位基因等情况),仅保留一个最好的组装结果;
- Chrysalis程序对组装得到的contig进行聚类,然后为每个类构建de Bruijn图。属于同一类的contig具有相似的序列,可能来源于可变剪切或旁系同源基因的独特部分;
- Butterfly程序分析reads与组装得到的contig的关系,报告所有可能的转录本序列,并解析可变剪接异构体以及旁系同源基因。
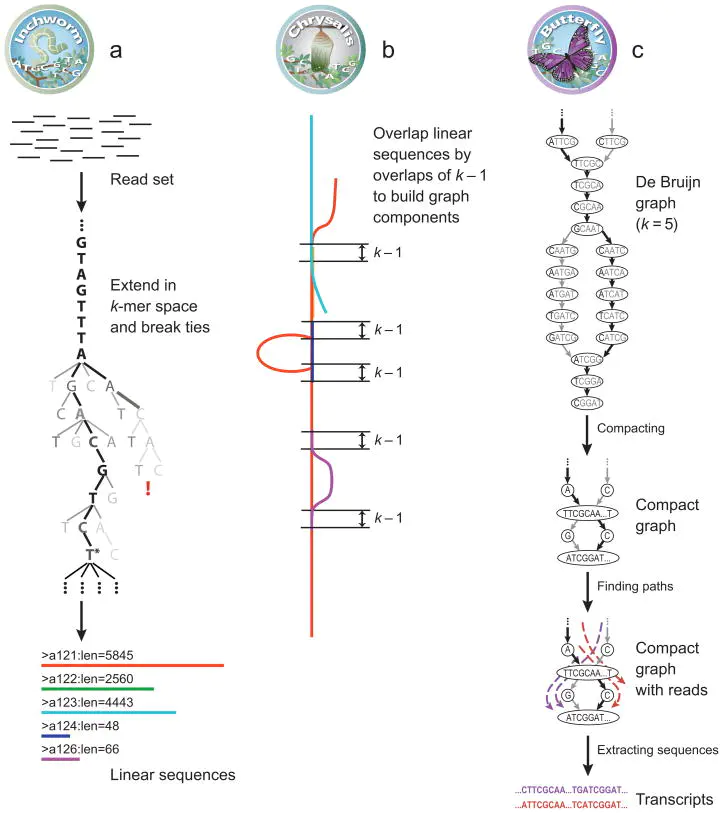
组装得到的基因、转录本数量和CG碱基比例如下表所示:
| 基因总数: | 96769 |
| 转录本(包含isoform)总数: | 141327 |
| CG碱基含量百分数: | 58.28 |
N50统计量(组装序列长度的中位数)常用来评估组装质量。然而简单使用N50统计量评估转录组的组装质量是不合适的,因为转录组组装的目的是恢复许多相对较短的contig,而不是少数非常长的contig,因此使用N50统计量会得到严重偏斜的组装结果统计。ExN50统计量是对传统N50统计量的修改,它避开了短的低表达转录本或长的高表达转录本的影响,使其适用于评估转录组组装。
我们可以依据Nx统计量对转录本进行组装质量评估,根据所有组装的contig计算得出N10 到 N50 值。
| Nx | Contig Number |
|---|---|
| Contig N10: | 4729 |
| Contig N20: | 3450 |
| Contig N30: | 2631 |
| Contig N40: | 2031 |
| Contig N50: | 1540 |
组装生成过多转录本同源异构体(isoform)时,contig N50值通常会被夸大。为了减轻这种影响,我们还将根据每个组装得到的gene的最长同源异构体来计算 Nx 值。
| Nx | Contig Number |
|---|---|
| Contig N10: | 4296 |
| Contig N20: | 3008 |
| Contig N30: | 2221 |
| Contig N40: | 1640 |
| Contig N50: | 1182 |
3.2.2 组装质量评估
为了确保组装转录本的质量能够用于后续分析,我们使用进行了转录本的质量评估。
评估转录组组装质量的一个指标是检查组装成全长(或接近全长)的转录本的数量。一个通用的评估方式是将组装的转录本与所有已知的蛋白质进行比对并统计匹配到蛋白质的数量。
Uniprot(Universal Protein)是包含蛋白质序列,功能信息,研究论文索引的蛋白质数据库。其中,Swiss-Prot旨在提供与高水平注释(例如,蛋白质功能,其域结构,翻译后修饰,变体等的描述)相关的可靠蛋白质序列,最小程度的冗余和高水平与其他数据库的集成级别。注释主要来自文献中的研究成果和E-value校验过计算分析结果。我们使用该数据库对组装得到的转录本进行分析,结果如下表所示。
| Hit Percent Cover Bin | Count in Bin | >Bin Below |
|---|---|---|
| 100 | 1240 | 1240 |
| 90 | 741 | 1981 |
| 80 | 611 | 2592 |
| 70 | 584 | 3176 |
| 60 | 578 | 3754 |
| 50 | 602 | 4356 |
| 40 | 673 | 5029 |
| 30 | 712 | 5741 |
| 20 | 660 | 6401 |
| 10 | 177 | 6578 |
同时,我们将reads回贴到转录本上,查看其比对率(通常应高于80%),结果如下表
| Sample | Overall Alignment Rate |
|---|---|
| test1 | 0.9831 |
| test2 | 0.9840000000000001 |
| test3 | 0.9847 |
| test4 | 0.9839 |
| test5 | 0.9843000000000001 |
| test6 | 0.9819 |
| test7 | 0.9828 |
| test8 | 0.9795 |
| test9 | 0.9840000000000001 |
3.3 组装转录本的注释
从头组装的序列本身并不包含注释信息。为了阐述这些转录本的生物学性质,需要对其结构、功能进行注释,并将其转化为人类可读的基因标签。
3.3.1 转录本的结构注释
为了从Trinity生成的de novo RNA-Seq转录组装序列中识别转录本序列中的候选编码区域,需要对转录本进行结构注释,这能够明确其功能区域和剪接模式,同时有助于减少组装转录组的冗余。
这里我们使用transDecoder进行结构注释,该软件主要包含以下内容:
- 在转录本序列中找到最小长度的开放阅读框架(ORF)。
- 计算与GeneID软件类似的对数似然分数 > 0。
- 当在第一个读框架中评分时,上述编码分数最高,与其他两个正向读框架中的分数相比。
- 如果发现一个候选ORF完全被另一个候选ORF的坐标所包含,则报告较长的那个。然而,单个转录本可以报告多个ORF(允许操纵子,嵌合体等)。
- 构建/训练/使用PSSM来优化起始密码子预测。
3.3.2 转录本的功能注释
对基因的功能注释通常依赖基因间的同源性。同源性(homology)指基因具有共同的祖先。按照产生方式不同,同源基因可分为直系同源(Ortholog),旁系同源(Paralogs),等等。其中,直系同源指不同物种中起源于它们最近的共同祖先(last
common ancestor,LCA)的同一个基因的不同基因,旁系同源指物种内部源于基因复制(gene
duplication)产生的不同基因。通常,同源基因具有序列和功能的相似性,而直系同源基因往往比旁系同源基因能保留更多的序列相似性和功能一致性[9]。通过识别不同物种间基因的同源关系,就可以使用已知功能的基因推断未知功能的基因。
由于识别直系同源关系比识别同源关系需要更多的计算资源和更加复杂的算法,大多数基因功能注释软件使用基于同源关系而不是基于直系同源关系的方法进行功能注释[10]。而eggnog-mapper软件能够有效识别直系同源关系,并使用直系同源关系进行基因功能注释[11],从而获得比BLAST,
InterProScan等更精准的注释。使用eggnog进行功能注释的过程可以分为以下三步:
- 序列映射步骤:使用HMMER或DIAMOND将每条待注释序列与eggnog数据库进行比对,找到它们对应的种子基因,从而确定待注释序列所属最佳的直系同源关系;
- 直系同源关系的确定:eggnog数据库预先构建了每组直系同源基因的系统发育关系。对每一条待注释序列,使用对应的种子序列和系统发育关系进行小范围直系同源关系的构建,同时进行删除亲缘关系较远的物种的基因等筛选;
- 功能注释:按照亲缘关系由进到远使用上述与待注释序列具有直系同源关系的基因的功能对待注释序列进行注释,注释内容包括Gene Ontology (GO) 条目,KEGG 通路,COG功能分类,及分配合适的基因名称(gene symbol)。
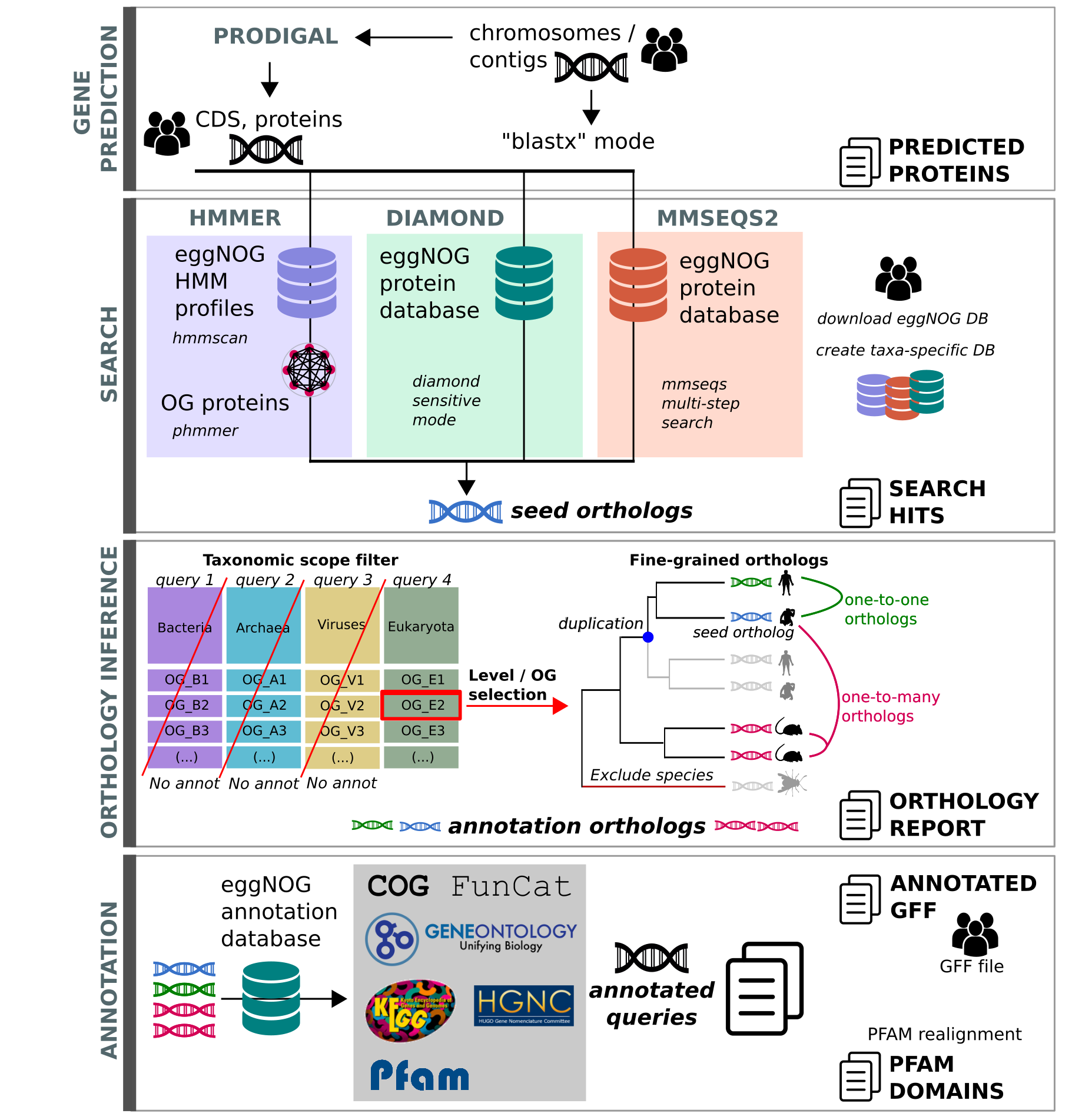
我们注释到以下数据库,
- query: trinity组装得到的基因ID
- seed_ortholog: 预测到的正交群(ortholog)的标识符
- evalue: 正交群与序列的比对期望值(E-value),用于评估比对结果的显著性
- score: 正交群与序列的比对得分,表示它们之间的相似程度
- eggNOG_OGs: Orthologous Group (OG) 号码,用于标识正交群
- max_annot_lvl: 与正交群关联的分类学信息,例如物种或分类单元
- COG: Clusters of Orthologous Groups (COG) 号码,用于描述基因和蛋白质的进化关系
- description: 序列的描述信息或注释概述
- Preferred_name: 既gene symbol
- GOs: Gene Ontology (GO) 注释,用于描述基因功能、细胞组分和生物过程的术语
- EC: Enzyme Commission (EC) 号码,用于标识酶的催化功能
- KEGG_ko: KEGG Orthology (KO) 标识符,用于标记与KEGG数据库中代谢通路和功能相关的基因
- KEGG_Pathway: 代谢通路的注释信息
- KEGG_Module: 代谢模块的注释信息
- KEGG_Reaction: 参与的生物反应的注释信息
- KEGG_rclass: 反应类别的注释信息
- BRITE: BRITE 层次结构的注释信息,用于描述生物实体的层次分类
- KEGG_TC: Transporter Classification (TC) 号码,用于标识膜转运蛋白的分类
- CAZy: 碳水化合物活性酶库(Carbohydrate-Active enZYmes)的注释信息,包括能催化碳水化合物降解、修饰、以及生物合成的相关酶系家族
- BiGG_Reaction: Biochemical, Genetic, and Genomic (BiGG) 数据库的注释信息
- PFAM是一个用于识别和注释蛋白质家族的数据库。它将具有相似结构和功能的蛋白质归类到相同的家族中
对常用的数据库COG_category,GOs,EC,KEGG_ko,CAZy,BiGG_Reaction,PFAMs注释信息进行统计,结果如下UpSet图所示。

3.3.3 KEGG和GO数据库的简介
KEGG(Kyoto Encyclopedia of Genes and Genomes)是用于生物系统表达和分析的数据库资源。KEGG主要使用通路图(Pathway Map)表示细胞和生物体的系统功能。通路图主要由表示基因和代谢物的节点和表示各类反应的边组成[12]。KEGG通路图最初的概念是通过手工创建以KEGG Orthology(KO)为代表的节点的泛型图,然后通过将KOs转换为特定的基因ID来适应每个有机体,从而产生有机体特异性的通路图。其中KO系统是一种将基因和蛋白质连接到KEGG通路图和其他分子网络的机制。每个KO都代表一组从实验中定义的、通用的直系同源基因,再通过手动和计算两种方式进行扩展,将特定生物中的实验知识推广到其他生物中[13]。
KEGG数据库包含15个依据前人发表的文献手动整理的部分如表所示:
| 分类 | 数据库名称 | 内容 | KEGG标识符 |
|---|---|---|---|
| 系统信息 | KEGG PATHWAY | 代谢通路图 | Map |
| 系统信息 | KEGG BRITE | BRITE 层次结构和表 | br/ko |
| 系统信息 | KEGG MODULE | 模块 | M |
| 基因组信息 | KEGG ORTHOLOGY(KO) | 直系同源基因 | K |
| 基因组信息 | KEGG GEONME | 物种基因组(包括一部分病毒基因组) | org code/T number |
| 基因组信息 | KEGG GENES | KEGG生物、病毒和附录类别的基因目录 | org:gene |
| 基因组信息 | KEGG SSDB | 条目之间的序列相似性 | |
| 化合物信息 | KEGG COMPOUND | 代谢物或其他小分子 | C number |
| 化合物信息 | KEGG GLYCAN | 多糖 | G number |
| 化合物信息 | KEGG REACTION | 生化反应 | R number |
| 化合物信息 | KEGG RCLASS | 反应类 | RC number |
| 化合物信息 | KEGG ENZYME | 酶的命名 | EC number |
| 健康信息 | KEGG DISEASE | 疾病 | H number |
| 健康信息 | KEGG DRUG | 药物 | D number |
| 健康信息 | KEGG DGROUP | 药物分类 | DG number |
| 健康信息 | KEGG ENVIRON | 天然药物和健康相关物质 | E number |
这里重点介绍与转录组相关的两个数据库:KEGG ORTHOLOGY和KEGG PATHWAY。
- 直系同源(Orthology)是指来源于被比较物种的最后一个共同祖先(last common ancestor, LCA)中同一个祖先基因的基因,随着物种形成事件(Speciation Events),祖先基因被不同的物种分别继承。由于具有同源性,直系同源基因在功能上往往是一致的,但并不绝对[8]。KEGG ORTHOLOGY数据库(KO)包含具有直系同源关系的基因(或编码蛋白质)信息及其所对应的功能。KO数据库中,直系同源基因以K开头。
- KEGG PATHWAY是KEGG数据库中最主要的部分,它储存着手动绘制的参考通路图。这些参考通路图由不同的物种特异性通路图依据基因间的直通同源关系计算生成,其中矩形代表直系同源基因而圆形代表代谢物。 每个通路的总图为map开头+五位数字编号(例如Map00020为TCA循环的总图),而物种代码+五位数字编号组成物种特异的通路图(例如hsa00020为人类的TCA循环通路。其中人类特有的直系同源基因用绿色表示)。 需要注意以下两组特殊的通路:Map01100(全局通路图)和Map01200(总览图),这两个通路分别代表代谢物的总体特征和主要特征,故不包含直系同源基因(矩形)。
基因本体 (Gene Ontology ,GO) 是生物学知识的结构化、标准化表示[14]。GO 描述通过正式定义的关系相互连接的概念 (也称为术语,或正式的类)。GO 被设计为物种不可知的,以便能够在整个生命树中对基因产物进行注释。GO 的计算框架能够实现一致的基因注释,跨生物体功能的比较,以及跨不同生物学数据库的知识整合。GO 包括以下三个方面:分子功能 (Molecular Function, MF)、细胞成分 (Cellular Component , CC) 和生物过程 (Biological Process, BP)[15]。
- 分子功能表示代表由基因产物执行的分子水平的反应。MFs 对应可以由单个基因产物 (即蛋白质或 RNA) 进行的反应,但是当某些反应不能被单个基因完成时,MF表示由基因复合体进行的反应。
- 细胞成分用于捕获发生分子功能的细胞位置。包括细胞解剖结构(cellular anatomical structures),基因表达产物所在的稳定的复合体或不属于细胞的病毒组分。
- 生物过程指由多种分子活性协同作用完成的较大过程或生物程序。
3.4 确定转录本的表达量
3.4.1 基于salmon的定量
Salmon 是一款用于 RNA测序(RNA-seq)数据转录本定量 的高效工具,主要用于估算基因或转录本的表达量(如 TPM、FPKM 或原始计数)。该软件采用准比对的策略,主要包含以下两个步骤:
- 准比对(Quasi-mapping)快速将 reads 匹配到参考转录组,记录可能的映射位置,但不保留详细比对信息。
- 丰度估计使用概率模型(如 变分推断 或 Gibbs采样)推断每个转录本的表达量,考虑多映射 reads 的分配问题(如一个 read 可能匹配多个同源转录本)。
各样本中不同基因表达量的分布箱线图
各样本中不同基因表达量百分比柱状图
3.4.2 样本间相关性分析
为了评估样本之间的相似性或差异性,验证实验设计的合理性,检测潜在异常样本或技术偏差(如批次效应),我们需要对不同样本的表达量数据进行相关性分析。不同生物学重复(相同处理组的样本)应呈现高相关性(Pearson/Spearman相关系数 >0.8),若相关系数过低,可能提示实验操作问题(如样本混淆、RNA降解或测序质量差等。相关性分析的结果见下图:
3.4.3 样本间主成分分析
主成分分析能够降维并揭示数据中的主要变异来源(如处理效应、批次效应或个体差异),直观展示样本分布模式,从而评估数据质量、验证实验假设。
3.4.4 不同基因的表达量热图
在RNA-seq或其他基因表达分析中,基因表达量热图(Heatmap) 是一种直观展示基因表达模式的可视化工具,常用于比较不同基因在不同样本或实验条件下的表达水平变化。
3.5 差异表达分析
为了寻找与特定生物学过程、表型或疾病相关的关键基因,需要对表达量数据进行差异表达分析(Differential Expression Analysis, DEA)。与基因芯片不同,二代测序转录组的表达量数据是离散型数据,因此不能直接使用基于正态分布的线性模型来建模。为了解释这种差异,已经有各种基于不同数据分布的算法和软件包被开发。这里我们默认使用limma软件包进行差异表达分析。 我们使用火山图(Volcano Plot)展示表达差异的结果。它结合统计学显著性和生物学效应大小,帮助快速识别显著差异表达的基因。在火山图中,横轴(X轴):通常为 log2 Fold Change(log2FC),表示基因表达量的变化倍数(处理组 vs 对照组)。 正值(右)代表基因在处理组中上调(如log2FC > 1,表示表达量翻倍); 负值代表基因在处理组中下调(如log2FC < -1,表示表达量减半); 而纵轴(Y轴通常为 -log10(p-value) 或 -log10(adjusted p-value),表示差异表达的统计学显著性,值越大(上):显著性越高(如 -log10(0.001)=3)。

3.6 功能富集分析
功能富集分析(Functional Enrichment Analysis) 是用于解释差异基因生物学意义的核心方法,通过识别这些差异基因在功能通路或生物过程中的聚集性,揭示其潜在的生物学机制。 我们使用两种方法(ORA,GSEA)分别对两个数据库(GO.KEGG)进行富集分析。得到以下结果
3.6.1 KEGG ORA

3.6.3 GO ORA

4 参考文献
- P. G. Higgs and N. Lehman, “The RNA World: molecular cooperation at the origins of life,” Nat. Rev. Genet., vol. 16, no. 1, pp. 7–17, Jan. 2015, doi: 10.1038/nrg3841.
- R. Stark, M. Grzelak, and J. Hadfield, “RNA sequencing: the teenage years,” Nat. Rev. Genet., vol. 20, no. 11, pp. 631–656, Nov. 2019, doi: 10.1038/s41576-019-0150-2.
- V. Raghavan, L. Kraft, F. Mesny, and L. Rigerte, “A simple guide to de novo transcriptome assembly and annotation,” Brief. Bioinform., vol. 23, no. 2, p. bbab563, Mar. 2022, doi: 10.1093/bib/bbab563.
- T. I. Garcia et al., “Effects of Short Read Quality and Quantity on a de novo Vertebrate Transcriptome Assembly,” Comp. Biochem. Physiol. Toxicol. Pharmacol. CBP, vol. 155, no. 1, pp. 95–101, Jan. 2012, doi: 10.1016/j.cbpc.2011.05.012.
- Chen S., Zhou Y., Chen Y., and Gu J., “fastp: an ultra-fast all-in-one FASTQ preprocessor,” Bioinformatics, vol. 34, no. 17, pp. i884–i890, Sep. 2018, doi: 10.1093/bioinformatics/bty560.
- S. W. Wingett and S. Andrews, “FastQ Screen: A tool for multi-genome mapping and quality control,” F1000Research, vol. 7, p. 1338, Sep. 2018, doi: 10.12688/f1000research.15931.2.
- P. E. C. Compeau, P. A. Pevzner, and G. Tesler, “How to apply de Bruijn graphs to genome assembly,” Nat. Biotechnol., vol. 29, no. 11, pp. 987–991, Nov. 2011, doi: 10.1038/nbt.2023.
- Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data - PMC.” Accessed: Aug. 30, 2024. [Online]. Available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3571712/
- Koonin E. V., “Orthologs, Paralogs, and Evolutionary Genomics,” Annu. Rev. Genet., vol. 39, no. 1, pp. 309–338, 2005, doi: 10.1146/annurev.genet.39.073003.114725.
- Huerta-Cepas J. et al., “Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper,” Mol. Biol. Evol., vol. 34, no. 8, pp. 2115–2122, Aug. 2017, doi: 10.1093/molbev/msx148.
- C. P. Cantalapiedra, A. Hernández-Plaza, I. Letunic, P. Bork, and J. Huerta-Cepas, “eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale,” Mol. Biol. Evol., vol. 38, no. 12, pp. 5825–5829, Dec. 2021, doi: 10.1093/molbev/msab293.
- M. Kanehisa and S. Goto, “KEGG: kyoto encyclopedia of genes and genomes,” Nucleic Acids Res., vol. 28, no. 1, pp. 27–30, Jan. 2000, doi: 10.1093/nar/28.1.27.
- M. Kanehisa, M. Furumichi, Y. Sato, Y. Matsuura, and M. Ishiguro-Watanabe, “KEGG: biological systems database as a model of the real world,” Nucleic Acids Res., p. gkae909, Oct. 2024, doi: 10.1093/nar/gkae909.
- Gene Ontology Consortium, “Gene Ontology Consortium: going forward,” Nucleic Acids Res., vol. 43, no. Database issue, pp. D1049-1056, Jan. 2015, doi: 10.1093/nar/gku1179.
- P. Gaudet and C. Dessimoz, “Gene Ontology: Pitfalls, Biases, and Remedies,” Methods Mol. Biol. Clifton NJ, vol. 1446, pp. 189–205, 2017, doi: 10.1007/978-1-4939-3743-1_14.