温馨提示:请使用火狐或者Chrome的网页浏览器来查看报告
微科盟宏基因组分箱数据分析结题报告
一 概述
宏基因组分箱(Binning)是将宏基因组测序得到的混合了 不同微生物的序列reads或序列组装得到的contigs或scaffolds按物种分开归类的过程。 这些分开归类的序列被称为宏基因组组装基因组(metagenome-assembled genomes,MAGs)。 传统的单物种全基因组序列都是经纯培养之后,再进行全基因组de novo测序才获得的, 但是环境中存在着大量的不可培养微生物(uncultured candidate bacterial species,未培养候选菌种), 而宏基因组分箱及相关技术不仅有助于获得不可培养微生物的基因组序列,还有以下诸多功能: (1)发现新物种,预测新物种基因,利用现有数据库分析新物种的功能; (2)扩充微生物基因组数据库,增加微生物多样性,提高宏基因组数据reads mapping率(检出率); (3)有助于宏基因组技术开发; (4)助力“感兴趣”微生物群结构和功能的研究; (5)为菌群的分类和功能描述提供了更多的解决方案; (6)通过 MAG GEM 分析, 指导设计培养基去培养一些具有特定功能的新菌株。 早在2011年,science上的一篇文章就用了宏基因组Binning技术对来自牛瘤胃的样本进行了宏基因组测序研究。 该研究从268 Gbp的宏基因数据中成功Binning出了15个不能培养的微生物的全基因组序列 [22]。
二 项目流程
2.1 实验流程
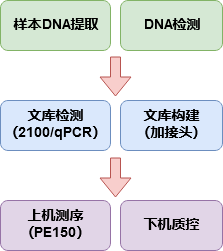
图2-1 测序流程图
2.2 数据分析流程
本报告中,宏基因组分箱的数据分析流程如图2-2标准分析部分所示, 主要步骤为质控,去宿主,分箱,MAG去冗余和筛选,MAG丰度计算(定量), MAG功能注释,MAG物种注释,MAG圈图等。
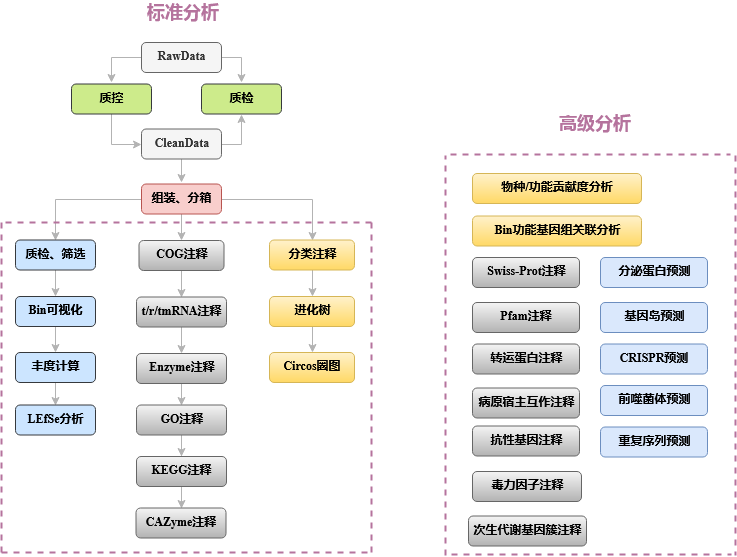
图2-2 宏基因组分箱数据分析流程
三 结果文件夹解读
00-QCStats序列质控和去宿主序列
|--00-QCStats
| |--1-QC_report_Rawfastq/*.html [原始数据fastqc质检结果]
| |--2-QC_report_Filtered/*.html [序列质控和去宿主序列之后的fastqc质检结果]
| |--reads_summary.txt [数据产出质量情况一览表]
本项目采用Illumina测序平台对测序样本进行双端测序。 基于FASTQ格式的测序文件是一种存储序列信息的特定文件,推荐用Notepad++等文本编辑器或者在电脑终端中打开。 FASTQ文件每四行对应一条测序Read:第一行以符号“@”起始,对应于序列ID和相应的描述信息; 第二行为实际测得的碱基序列;第三行以符号“+”起始; 而第四行的字符串则记录了第二行序列中每个碱基所对应的测序质量。
采用 Illumina测序平台测序获得的原始数据(Raw Data)存在一定比例低质量数据,
为了保证后续分析的结果准确可靠,需要对原始的测序数据进行预处理,包括质控
(Trimmomatic[20]
参数:ILLUMINACLIP:adapters_path:2:30:10 SLIDINGWINDOW:4:20 MINLEN:50),
和去宿主序列(Bowtie2[21]参数:--very-sensitive),获取用于后续分析的有效序列(clean data)。
测序数据预处理统计结果见表 3-1。序列质控步骤关键参数解释如下:
- 去除接头序列 (参数ILLUMINACLIP:adapters_path:2:30:10);
- 扫描序列(4bp滑窗大小),如果平均质量分低于20(99%正确率),切除后续序列(参数SLIDINGWINDOW:4:20);
- 去除最终长度小于50bp的序列(参数MINLEN:50)。
表3-1 数据产出质量情况一览表
| Sample ID | InsertSize(bp) | SeqStrategy | RawReads(#) | Raw Base(GB) | %GC | Raw Q20(%) | Raw Q30(%) | Clean Reads(#) | Cleaned(%) | Clean Q20(%) | Clean Q30(%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 | 350 | (150:150) | 23613034 | 7.08 | 61 | 97.70 | 93.78 | 22416119 | 94.93 | 98.80 | 95.62 |
| C2 | 350 | (150:150) | 27584954 | 8.28 | 61 | 97.10 | 92.62 | 26060963 | 94.48 | 98.55 | 94.94 |
| C3 | 350 | (150:150) | 23777022 | 7.13 | 61 | 98.45 | 95.71 | 22889304 | 96.27 | 99.27 | 97.08 |
| C4 | 350 | (150:150) | 27007788 | 8.10 | 61 | 97.63 | 93.65 | 25636029 | 94.92 | 98.77 | 95.52 |
| T11 | 350 | (150:150) | 29809022 | 8.94 | 62 | 97.62 | 93.48 | 28346006 | 95.09 | 98.63 | 95.16 |
| T12 | 350 | (150:150) | 24702558 | 7.41 | 61 | 97.72 | 93.78 | 23570823 | 95.42 | 98.72 | 95.43 |
| T13 | 350 | (150:150) | 28274974 | 8.48 | 61 | 97.63 | 93.54 | 26703496 | 94.44 | 98.69 | 95.32 |
| T21 | 350 | (150:150) | 25016746 | 7.51 | 60 | 97.74 | 93.74 | 23831131 | 95.26 | 98.70 | 95.35 |
| T22 | 350 | (150:150) | 24980036 | 7.49 | 60 | 97.55 | 93.21 | 23555813 | 94.30 | 98.57 | 94.96 |
| T23 | 350 | (150:150) | 24026221 | 7.21 | 62 | 97.64 | 93.58 | 22758509 | 94.72 | 98.69 | 95.34 |
- Sample ID: 样品名;
- InsertSize(bp): InsertSize是在建库切胶时选择的长度,合适的InsertSize能避免测序接头污染;
- SeqStrategy: 测序策略(一般为双端,各150bp);
- RawReads(#):测序Raw reads的数量;
- Raw Base(GB): 以GB为单位的Raw reads数量, 测序原始数据的总碱基数,即为Raw reads数量乘以测序长度算得;
- %GC:G/C碱基数占总碱基数量的百分比;
- Clean Reads(#):过滤(质控和去宿主序列)后获得的Clean reads 数量;
- Cleaned(%):过滤后剩余的序列数占Raw reads 的百分比;
- Q20: 质量分高于20的碱基所占比例;
- Q30,质量分高于30的碱基所占比例。
01-MAG分箱与MAGs质控
文件说明: |--01-MAG | |--1-MAG_raw/*.fa [所有分组分箱得到的原始MAGs] | |--2-MAG_pick/*.fa [筛选、去冗余、重组装后的最终MAGs] | |--MAGs_picked_summary.txt [最终MAGs统计:完整度、污染度、N50、N90、Size、GC含量] | |--checkm_raw.txt [所有原始MAGs的完整度与污染度评估]
分箱(Binning)分析步骤如下(除非特别说明, 否则各软件使用默认参数):
- 混合组装: 运用Megahit[1] 将同一分组内各样本的clean reads列表作为输入 进行组装,得到分组contigs。
- 建索引与序列比对:运用BWA[3] 对分组contigs建索引, 将各样本的clean reads比对至对应分组contigs; 运用Samtools[4]对比对结果排序并建立索引,生成BAM文件。
- 混合分箱: 基于BAM文件计算分组contigs的多样本 测序深度(depth,分箱领域,有时也叫coverage), 挑选各分组长度大于1000bp的contigs(MetaBAT2除外, 它强制要求contig长度>1500bp), 运用MetaBAT2[5] 对各分组的contigs进行分箱,获得原始MAGs。
- MAGs提纯:运用RefineM[9] 对MAGs进行进一步提纯,去除其中污染度较高的contigs。
- 质量评估与去冗余:合并所有分组的MAGs, 运用CheckM[10]的lineage_wf流程评估MAGs的完整度和污染度; 运用dRep[11]对MAGs进行去冗余 (dRep参数:--P_ani 0.9 --S_ani 0.95 --completeness 90 --contamination 10) ,得到去冗余后的 MAGs 集合。
- 重组装:针对上一步去冗余后的 MAGs,根据前面“建索引与序列比对”步骤的比对结果BAM文件, 以及各 MAG 的contigs集合信息,提取各MAG对应的特异reads, 运用MetaWRAP[8]的reassemble_bins模块 (基于SPAdes[12])对各MAG进行重组装。 重组装完成后,再次运行 CheckM[10] 评估这些最终 MAGs 的完整度和污染度。 表3‑2 展示的即为重组装后最终 MAGs 集合的质量统计;与去冗余阶段的结果(checkm_raw.txt)相比, 重组装常能使部分 MAGs 的完整度提高、污染度降低。
表3-2 最终MAGs统计
| MAGID | Completeness | Contamination | N50 | N90 | Size | GC_percent |
|---|---|---|---|---|---|---|
| MAG.C.10 | 94.94 | 5.454 | 31747 | 6141 | 3462104 | 0.520942 |
| MAG.C.6 | 98.94 | 2.350 | 70535 | 8431 | 2653583 | 0.464926 |
| MAG.T1.102 | 99.01 | 1.642 | 122765 | 31767 | 3203337 | 0.427849 |
| MAG.T1.117 | 93.05 | 0.925 | 128251 | 37877 | 4288915 | 0.595889 |
| MAG.T1.121 | 94.49 | 0.628 | 24345 | 4693 | 3742350 | 0.613720 |
| MAG.T1.124 | 94.81 | 1.272 | 44935 | 8666 | 3549206 | 0.423166 |
| MAG.T1.126 | 93.19 | 0.246 | 74352 | 21536 | 2635269 | 0.477953 |
| MAG.T1.12 | 94.27 | 0.911 | 117523 | 21375 | 3867357 | 0.681245 |
| MAG.T1.161 | 95.22 | 0.654 | 106859 | 25922 | 3249281 | 0.430625 |
| MAG.T1.171 | 91.55 | 1.485 | 41209 | 9892 | 4884817 | 0.495622 |
| MAG.T1.179 | 91.97 | 3.963 | 49965 | 9674 | 3365536 | 0.725438 |
| MAG.T1.194 | 94.58 | 1.724 | 53925 | 10629 | 3497051 | 0.489122 |
| MAG.T1.209 | 97.21 | 1.149 | 47470 | 13034 | 6164932 | 0.530426 |
| MAG.T1.20 | 90.77 | 2.717 | 21734 | 5652 | 2601952 | 0.613953 |
| MAG.T1.22 | 95.94 | 1.700 | 79118 | 24891 | 6209475 | 0.611487 |
| MAG.T1.36 | 90.92 | 2.384 | 35457 | 7857 | 4052318 | 0.623586 |
| MAG.T1.43 | 96.52 | 3.300 | 58296 | 15357 | 3000518 | 0.382869 |
| MAG.T1.58 | 91.36 | 2.721 | 31084 | 7523 | 5980521 | 0.664247 |
| MAG.T1.5 | 96.76 | 3.181 | 278228 | 63381 | 4532866 | 0.592558 |
| MAG.T1.71 | 100.00 | 0.026 | 89356 | 24678 | 4372432 | 0.387973 |
| MAG.T1.77 | 93.81 | 3.681 | 12218 | 3684 | 2891395 | 0.339671 |
| MAG.T1.84 | 98.67 | 0.000 | 168569 | 47359 | 3661158 | 0.454820 |
| MAG.T2.152 | 92.58 | 1.275 | 221551 | 76508 | 2174759 | 0.637379 |
| MAG.T2.180 | 94.06 | 0.656 | 130680 | 25341 | 3142183 | 0.372610 |
| MAG.T2.198 | 99.70 | 0.000 | 214598 | 79632 | 4044987 | 0.421501 |
| MAG.T2.204 | 96.98 | 3.932 | 66500 | 16432 | 4580347 | 0.647177 |
| MAG.T2.27 | 99.24 | 0.187 | 196540 | 52784 | 4899526 | 0.463914 |
| MAG.T2.32 | 97.40 | 3.095 | 51482 | 12196 | 3262439 | 0.368905 |
| MAG.T2.42 | 94.93 | 4.204 | 17379 | 5261 | 4365942 | 0.702159 |
| MAG.T2.48 | 97.15 | 1.136 | 257831 | 73593 | 3642140 | 0.642161 |
| MAG.T2.55 | 93.25 | 3.866 | 54936 | 13970 | 6957463 | 0.540898 |
| MAG.T2.62 | 94.29 | 3.378 | 39867 | 12293 | 3461391 | 0.603699 |
- 第一列:MAG编号;
- 第二列:MAG完整度;
- 第三列:MAG污染度;
- 第四列:Contig N50是指将一个MAG的Contig按照从大到小的顺序依次相加, 当相加的长度达到Contig总长度的一半时,最后一个加上的Contig长度;
- 第五列:Contig N90是指将一个MAG的Contig按照从大到小的顺序依次相加, 当相加的长度达到Contig总长度的90%时,最后一个加上的Contig长度;
- 第六列:一个MAG的所有Contig长度之和,即该基因组草图的总长度;
- 第七列:GC含量是指一个MAG中GC碱基占总碱基的比例。
02-MAG_Plot 分箱效果散点图
文件说明: |--02-MAG_Plot | |--MAGs_contig_summary.txt [散点图输入文件,含每个MAG重组装前的contig的GC含量和contig的深度信息] | |--MAGs_contig_summary.svg [绘图结果,利用GC和depth数据把每个MAG重组装前的contig 画到图中]
利用可视化的方法直观展示MAG的由来和所含信息。 宏基因组分箱的原理是根据序列(重组装前contig/scaffold) 四核苷酸频率和序列丰度变化模式将序列分成一个个MAGs。 每个MAG的每个contig都会在此处理(分箱)的过程中有一个depth深度数据。 另外我们利用自己的python脚本计算每个MAG的contig GC含量。 有了contig GC含量和depth数据即可进行MAG可视化,绘制每个MAG中每个contig的散点图(图3-1)。

图3-1 Binned contigs(重组装前)可视化
横坐标是contig的GC含量;纵坐标是contig depth; 一个点代表一个contig,相同颜色的contig来自同一个MAG。
03-MAG_Abundance 丰度分析
03-MAG_Abundance ├── 1-Barplots [丰度柱形图] ├── 2-Heatmaps [丰度热图] ├── 3-Circos [丰度圈图] ├── 4-SignificanceAnalysis [丰度组间显著性差异比较] ├── 5-CorrelationAnalysis [丰度相关性分析] └── MAGs_abundance_table.xls [MAGs在各样本的丰度汇总表]
使用MetaWRAP[8] 的quant_bins模块(Salmon算法)计算每个MAG的丰度。 原理是将样本clean reads比对到MAG中的contigs, 计算单个样本某个MAG中所有contigs的总TPM。
我们用R语言pheatmap绘图函数绘制样品-MAG丰度热图。 热图可以以色块颜色深浅的方式表达MAG丰度的大小,还可以进行MAG-MAG聚类和样品-样品聚类,相似的丰度模式会被聚到一起。 然后,统计每个MAG的丰度总数,用ggplot geom_bar绘制柱形图并排序,展示整批数据中所有MAG的丰度情况。 如果客户提供的样品分组>=2且每组样品在三个以上,那么还可以用lefse进行组间差异分析寻找与分组有关的MAG。
1-Barplots文件夹中包含按照分组顺序排列的每个样本的MAG丰度柱形图(图3-2),以及计算分组均值后画的柱形图。

图3-2 丰度柱形图
横坐标(Sample Name)是样品名,纵坐标(Sequence Number Percent)表示MAG的丰度占总丰度的比率,柱状图自上而下的颜色顺序对应于右侧的图例颜色顺序。图例中最多显示最优势的20个MAGs,余下的相对丰度较低的MAG被归类为Other在图中展示。
2-Heatmaps文件夹中包含按照分组顺序排列的每个样本的MAG丰度热图(图3-3),以及计算分组均值后画的热图。

图3-3 丰度聚类热图
说明:纵轴为样品名称信息,同时也包括了分组信息。横轴为MAG ID。图中上方的聚类树为MAG在各样本中丰度分布的相似度聚类,左侧的聚类树为样品聚类树,中间的热图是MAGs的相对丰度(log10(TPM))热图,颜色与相对丰度的关系见图上方的刻度尺。
3-Circos 文件夹中包含按照分组顺序排列的每个样本的MAG丰度Circos图(图3-4),用于展示每个样本各个MAG(丰度前10)的丰度比例,以及每个MAG在各个样本中的丰度比例。

图3-4 丰度Circos图
说明:左半圈为丰度最高的十个MAG,每个MAG内,不同颜色代表不同样本来源的丰度比例;右边半圈为样本,每个样本内不同颜色代表不同MAG的丰度比例。
4-SignificanceAnalysis文件夹中, DunnTest文件夹包含了DunnTest组间多重比较结果表格,第一列为MAG ID, KW_pvalue是Kruskal-Wallis算出总的p值,DunnTest_comparison是比较的分组对, DunnTest_Z是DunnTest检验统计量,DunnTest_PValueAdjusted是Bofferoni校正错误发现率后的DunnTestp值; LEfSe文件夹中包含了LEfSe分析[23]中, LDA分值大于2的MAGs柱形图(图3-5), LEfSe寻找每一个分组的特征MAGs(默认为LDA>2的MAG), 也就是相对于其他分组,在这个组中丰度较高的MAG。

图3-5 LEfSe分析LDA柱形图
说明:每一横向柱形体代表一个MAG,柱形体的长度对应LDA值,LDA值越高则分组间该MAG的丰度差异越大。柱形的颜色对应该MAG是那个分组的特征MAG(在对应分组中的丰度相对较高)。
只有提供了环境因子,5-CorrelationAnalysis中才会包含内容。其中,CorrelationHeatmap文件夹中包含了相关性热图(图3-6);RDA文件夹中包含了RDA分析结果图(图3-7)。CCA/RDA的分析主要依赖R语言VEGAN包,以及用ggplot2进行可视化。CCA/RDA(DCA判断用哪一种分析)分析是基于对应分析发展的一种排序方法,将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析。RDA是基于线性模型,CCA是基于单峰模型。本报告先进行DCA分析,看最大轴的值是否大于4,如果大于4.0,就选CCA,否则选RDA。该分析主要用来反映菌群与环境因子之间的关系,可以检测环境因子、样品、菌群(抗性基因,KEGG功能)三者之间的关系或者两两之间的关系,可得到影响样品分布的重要环境驱动因子。该分析给出的所有p值都是反映解释变量(连续的数值变量,或者分类变量)对MAGs群落丰度结构的解释程度是否显著(简单的说就是解释变量对MAGs 丰度是否有影响,影响是否显著),所有p值都是用R语言VEGAN包里的置换检验得出的(permutation test),*_features_location_plot图中(图3-7)的p值反映了所有连续的数值变量(环境因子)对MAGs丰度差异的解释程度(总的p值),表格*_RDA.envfit中的p值反映了每个环境因子对MAGs丰度差异的解释程度;*_RDA_sample_location_plot图中的p值反映了分组对MAGs丰度差异的解释程度,p<0.05,解释方差显著,图中样本点之间的距离近似于样本之间MAGs丰度结构差异程度,样本点投影到环境因子对应的值近似于该样本真实的环境因子值。

图3-6 MAGs与环境因子之间的相互关系热图
说明:X轴上为环境因子,Y轴为MAGs。计算获得R值(秩相关)和校正错误发现率的P值。R值在图中以不同颜色展示,右侧图例是不同R值的颜色区间。* 0.01≤ P <0.05,** 0.001≤P < 0.01,*** P < 0.001。

图3-7 MAGs CCA/RDA排序图
说明:在CCA/RDA MAGs排序图内,环境因子用箭头表示, 箭头连线的长度代表某个环境因子与MAGs丰度分布间相关程度的大小(解释方差的大小), 箭头越长,说明相关性越大,反之越小。箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小, 夹角越小,相关性越高;反之越低。 环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系,钝角时呈负相关关系。 每个点代表一个MAG,点越大,对应MAG丰度越高, 灰色点代表丰度较低的MAGs,未在图中注释MAGs名称,将MAGs投影到各个环境因子, 对应的值即为该MAGs倾向于存在的环境(喜欢的环境)。或者说,将MAGs点与原点连线,MAGs间, MAGs与环境因子间,环境因子间的夹角的余弦值近似于相关系数,至于有多近似, 就要看RDA1/CCA1和RDA2/CCA2两个坐标轴的解释方差百分比有多大,越大越近似。
04-MAG_Function MAGs功能注释情况统计
04-MAG_Function [功能注释结果文件夹] ├── CAZyme [CAZy碳水化合物数据库注释结果] ├── COG [eggNOG数据库COG注释结果] ├── GO [GO数据库注释结果] ├── KEGG [KEGG数据库注释结果] └── * ├── *_detail_MAG [包含每个MAG 注释到数据库基因家族的种类和CDS个数统计图] | └── *_annotations.txt [每个MAG的原始注释结果文件] ├── *_detail_MAG_L1 [包含每个MAG 注释到数据库基因家族level1水平的种类和CDS个数统计图] ├── *_detail_gene [包含每个注释到的数据库基因家族来源于各个MAG 的CDS个数统计图] ├── *_summary [注释情况汇总统计图,由于图片大小限制,只会展示CDS个数排名靠前的基因家族和MAGs] ├── *_GeneCount.xls [MAGs注释到数据库基因家族的CDS个数统计表] └── *_GeneCount.L1.xls [MAGs注释到数据库基因家族level1水平的CDS个数统计表]
使用Prokka软件[13] 对每个MAG进行功能注释,Prokka是常用的基因组注释软件, Prokka先使用Prodigal[14] 预测得到每个MAG中的基因, 得到核苷酸序列(基因序列)和氨基酸序列 (通过核苷酸序列翻译得到,也常称为蛋白序列),将序列与已有数据库信息比对, 获得每个MAG中的rRNA、tRNA、tmRNA、CDS、直系同源蛋白簇(COG)、EC等注释信息。
我们接下来对Prokka得到的每个MAG中的氨基酸序列做进一步注释。 使用KofamKOALA[15] 的离线版本Kofamscan进行KEGG功能注释。 使用emapper[16] 注释得到多个数据库的注释信息(包括GO)。 使用Diamond软件[17] 比对CAZy数据库, 对每个MAG进行碳水化合物酶注释, 获取每个MAG中的碳水化合物酶信息。
有了每个MAG中的基因组注释信息,我们就可以统计每个基因组(MAG)上各个基因家族的数目 (同一个基因家族在该基因组上注释到的次数,可理解为拷贝数)。
04-MAG_Function文件夹包含了各个功能数据库的注释情况统计;接下来我们将对每个功能数据库展开说明:
04-MAG_Function/CAZyme CAZy碳水化合物数据库注释
碳水化合物酶(CAZy)数据库是关于能够合成或者分解复杂碳水化合物和糖复合物的酶类的数据库。CAZy数据库基于蛋白质结构域中的氨基酸序列相似性将碳水化合物活性酶类归入不同蛋白质家族。CAZy数据库提供了酶分子序列的家族信息,物种来源,基因序列,蛋白序列,三维结构,EC分类,相关数据库链接。此数据库可以将酶分子的序列、结构、催化机制关联起来。CAZy有三个level,第一个level是六大功能类,即GH GT AA CE PL CBM;第二个level是CAZy family;第三个是有EC编号的具体酶信息,如EC2.4.1.129。
利用蛋白比对工具diamond将prokka预测得到的所有功能区(CDS)与CAZy数据库进行比对可获取基因组CAZyme family注释信息。
碳水化合物活性酶数据库中CAZymes功能分类如下:
| 名称 | 缩写 |
|---|---|
| 糖苷水解酶类 | GHs |
| 糖苷转移酶类 | GTs |
| 多糖裂解酶类 | PLs |
| 糖水化合物脂酶类 | CEs |
| 碳水化合物结合模块 | CBMs |
| 辅助模块酶类 | AAs |
部分结果统计图如下:

图3-8 Level1 CAZymes数量统计箱图
说明:横坐标是 CAZymes的level1大类,纵坐标是每一类CAZyme在各个MAG中的CDS数量, 每个箱子的高度表示CAZy数量的变化幅度,箱子的首和尾是分位数,中间的横线是中位数。

图3-9 Level1 CAZymes数量统计热图
说明:pheatmap热图可以展示每个MAG的CAZyme CDS计数,不同MAG之间的相似度(横向)和不同CAZyme类别之间的相似度(纵向)。图中每一个色块表示一个MAG中的一类CAZyme CDS计数,颜色越蓝表示个数越低,越红表示个数越高。

图3-10 Level1 CAZymes数量统计堆叠图
说明:一种颜色表示一类CAZyme;色柱的高度表示该CAZy的CDS计数

图3-11 Level1 CAZymes数量统计饼图
说明:该饼图展示每个MAG中的每一个CAZyme类别CDS计数与该MAG中总数量的比例。

图3-12 某个CAZyme 在每个MAG中的CDS数量统计饼图
说明:图片篇幅限制,只展示CDS数量排名前10的MAGs
04-MAG_Function/COG COGs注释
COG,即Clusters of Orthologous Groups of proteins(同源蛋白簇)。COG是由NCBI创建并维护的蛋白数据库,根据细菌、藻类和真核生物完整基因组的编码蛋白系统进化关系分类构建而成。COG分为两类,一类是原核生物的(一般称COG),另一类是真核生物(一般称KOG)。通过比对可以将某个蛋白序列注释到某一个COG中,每一簇COG由直系同源序列构成,从而可以推测该序列的功能。
图3-13 展示了COG level2水平其中一个MAG的CDS计数统计柱形图,其它MAGs请查看结果文件夹。

图3-13 COG数据库分类注释统计图
说明:该图描述的是预测基因(CDS)注释到COG不同分类水平数量情况;纵坐标是COG level 2分类(字母表示,共26种分类);横坐标是注释到的CDS计数;颜色是COG level 1分类(四大类,分别是:细胞过程和信号传递、信息储存和加工、代谢和尚未明确);柱子越长,属于该分类的预测基因越多。
COG level 2分类的26个字母的意义如下:
| COG Function Category | Level 1 | Level 2 |
|---|---|---|
| J | INFORMATION STORAGE AND PROCESSING | Translation, ribosomal structure and biogenesis |
| A | INFORMATION STORAGE AND PROCESSING | RNA processing and modification |
| K | INFORMATION STORAGE AND PROCESSING | Transcription |
| L | INFORMATION STORAGE AND PROCESSING | Replication, recombination and repair |
| B | INFORMATION STORAGE AND PROCESSING | Chromatin structure and dynamics |
| D | CELLULAR PROCESSES AND SIGNALING | Cell cycle control, cell division, chromosome partitioning |
| Y | CELLULAR PROCESSES AND SIGNALING | Nuclear structure |
| V | CELLULAR PROCESSES AND SIGNALING | Defense mechanisms |
| T | CELLULAR PROCESSES AND SIGNALING | Signal transduction mechanisms |
| M | CELLULAR PROCESSES AND SIGNALING | Cell wall/membrane/envelope biogenesis |
| N | CELLULAR PROCESSES AND SIGNALING | Cell motility |
| Z | CELLULAR PROCESSES AND SIGNALING | Cytoskeleton |
| W | CELLULAR PROCESSES AND SIGNALING | Extracellular structures |
| U | CELLULAR PROCESSES AND SIGNALING | Intracellular trafficking, secretion, and vesicular transport |
| O | CELLULAR PROCESSES AND SIGNALING | Posttranslational modification, protein turnover, chaperones |
| X | CELLULAR PROCESSES AND SIGNALING | Mobilome: prophages, transposons |
| C | METABOLISM | Energy production and conversion |
| G | METABOLISM | Carbohydrate transport and metabolism |
| E | METABOLISM | Amino acid transport and metabolism |
| F | METABOLISM | Nucleotide transport and metabolism |
| H | METABOLISM | Coenzyme transport and metabolism |
| I | METABOLISM | Lipid transport and metabolism |
| P | METABOLISM | Inorganic ion transport and metabolism |
| Q | METABOLISM | Secondary metabolites biosynthesis, transport and catabolism |
| R | POORLY CHARACTERIZED | General function prediction only |
| S | POORLY CHARACTERIZED | Function unknown |
04-MAG_Function/GO GO注释
GO数据库是基因本体联合会(Gene Onotology Consortium)所建立的数据库,旨在建立一个适用于各种物种的,对基因和蛋白质功能进行限定和描述的,并能随着研究不断深入而更新的语言词汇标准。GO是多种生物本体语言中的一种,是OBO(Open BiomedicalOntologies)组织中的一员,GO提供了一系列的语义(terms)用于描绘基因、基因产物的特点,这些语义通过三个概念维度展开:细胞学组件(Cellular Component)用于描述某个节点的亚细胞结构、位置和大分子复合物,如外部封装结构(external encapsulating structure)等;分子功能(molecular function),用于描述基因以及基因产物的功能,比如蛋白质结合转录因子活性(protein binding transcription factor activity);生物学途径(biological process)指的是分子功能的有序组合以实现更复杂的生物功能,例如树突状细胞的抗原处理和提呈(dendritic cell antigen processing and presentation)。
使用eggnog-mapper功能注释软件和eggNOG数据库进行GO注释。该方法除了获得GO的注释信息还能获得KEGG、COG等信息。获得GO结果后统计注释到每个GO中的基因数量,然后进一步归类获得GO的category分类信息。最后通过R语言对该数据进行柱形图绘制。
图3-14 展示了其中一个MAG预测基因所属GO的计数,其它MAGs请查看结果文件夹。

图3-14 GO注释统计图
该图说明的是其中一个MAG预测基因所属GO的情况;横坐标是数量;纵坐标是GO;颜色是GO分类;柱子越高,该GO中包含的预测基因(CDS)越多。
04-MAG_Function/KEGG KEGG注释
KEGG数据库于 1995 年由 Kanehisa Laboratories 推出 0.1 版,目前发展为一个综合性数据库,其中最核心的为 KEGG PATHWAY 和 KEGG ORTHOLOGY 数据库。在 KEGG ORTHOLOGY 数据库中,将行使相同功能的基因聚在一起,称为 Ortholog Groups (KO entries),每个 KO 包含多个基因信息,并在一至多个 pathway 中发挥作用。而在 KEGG PATHWAY 数据库中,将生物代谢通路划分为 6 类,分别为:细胞过程(Cellular Processes)、环境信息处理(Environmental Information Processing)、遗传信息处理(Genetic Information Processing)、人类疾病(Human Diseases)、新陈代谢(Metabolism)、生物体系统(Organismal Systems),其中每类又被系统分类为二、三、四层。第二层目前包括有 57个种子 pathway;第三层即为其代谢通路图;第四层为每个代谢通路图的具体注释信息。
使用KEGG注释软件KofamScan和KOfam数据库对prokka预测基因的蛋白序列进行KEGG注释。其中一个MAG的KEGG pathway level2水平的CDS数量如图3-15所示,其它MAGs请查看结果文件夹。

图3-15 KEGG pathway level2水平CDS数量可视化
横坐标是KEGG level2水平CDS数量;纵坐标是KEGG level 2名称;颜色用于区分KEGG level 1的类型。
KEGG数据库有比较详细的通路图,我们将每个MAG注释到的KO(KEGG Ortholog Groups)在通路图中高亮显示。其中一个MAG的其中一个通路如图3-16所示,其它结果请看结果文件夹。
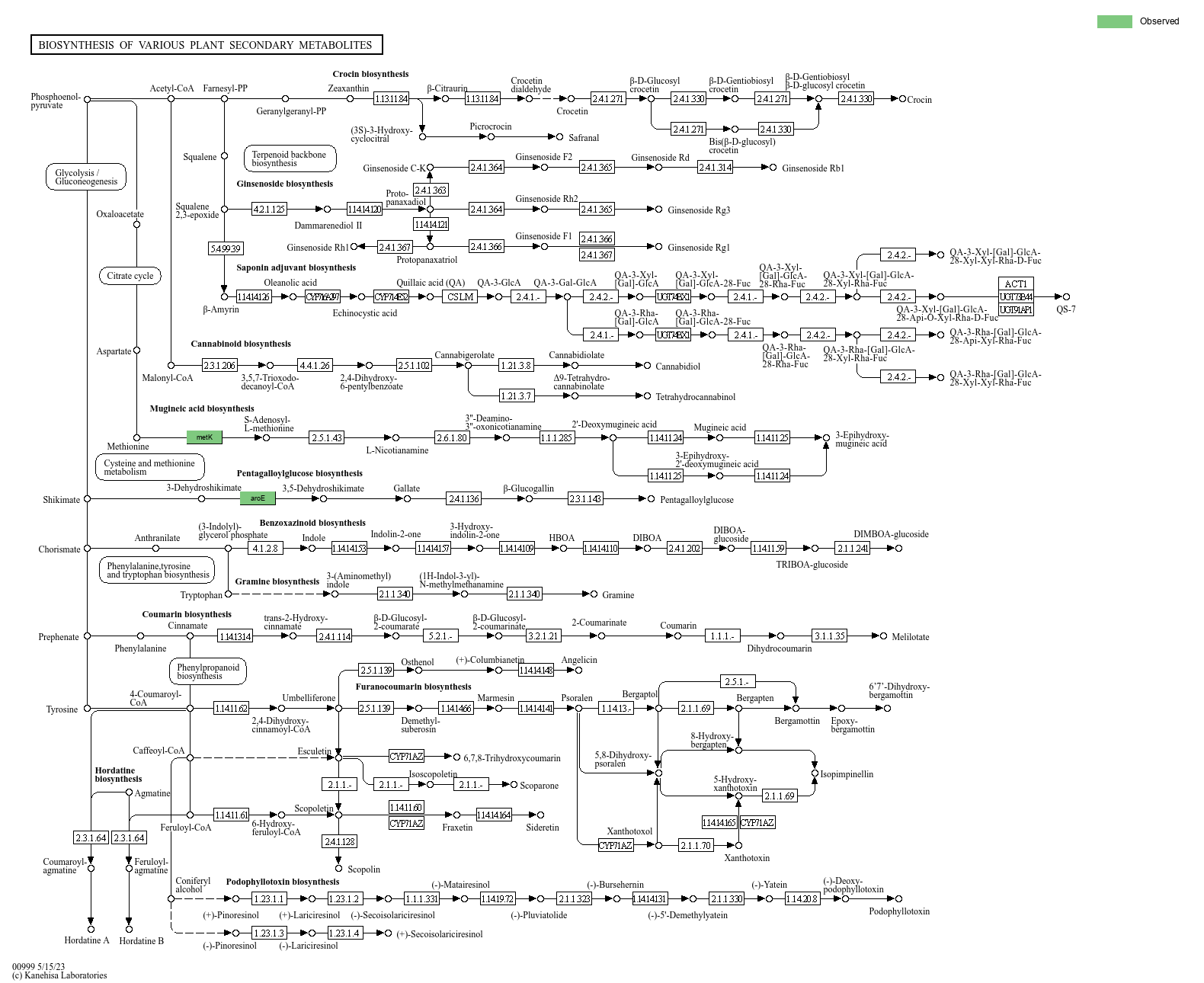
图3-16 MAG 注释到的基因通路图
高亮显示的基因是在该MAG中注释到的基因。对应的KO ID可以在相应的网页中查看(打开map*.html,鼠标悬浮在色块上即可)
KEGG通路的符号图例如下:

05-MAG_Taxonomy MAGs物种注释和进化分析
05-MAG_Taxonomy/ [MAGs物种注释和进化分析结果文件夹] ├── MAGs_abundance_taxonomy.xls [MAGs丰度表,同时在最后一列加上了MAG的物种分类注释] ├── MAGs_tree.tre [MAGs 进化树,nwk格式] ├── MAGs_taxonomy_detail.xls [物种注释的详细情况统计] ├── MAGs_novel_species.xls [根据GTDB-Tk分类结论推断的可能的新物种MAGs列表] ├── phylogenetic_tree_heatmap_ID.pdf [进化树可视化,标注MAG ID] ├── phylogenetic_tree_heatmap.pdf [进化树可视化,标注MAG 所属的属水平分类] └── phylogenetic_tree_table.xls [与进化树中的热图对应的丰度表]
PhyloPhlAn是发表在Nature Communications上的一个用于分析微生物之间进化关系的软件, PhyloPhlAn3在PhyloPhlAn基础上作了很多改进,再次发表于Nature Communications [18]。 我们使用PhyloPhlAn3软件,基于Prokka得到的MAG中的氨基酸序列, 通过分析400多种通用微生物蛋白, 计算MAGs之间的进化树。
使用GTDB-Tk软件[19](默认参数), 将MAGs(基因组)与GTDB-Tk软件配套的 GTDB数据库比较(Release 220), 获取每个MAG的物种分类信息。
我们可以将多序列比对覆盖率 (msa_percent) > 80%, 同时GTDB-Tk分类结论为“taxonomic novelty determined using RED” 或“Genome not assigned to closest species as it falls outside its pre-defined ANI radius” 的MAGs推断为新物种(MAGs_novel_species.xls表)
图3-17是MAG之间的进化关系树状图,图中的热图展示的是MAG丰度信息,树状图由R语言ggplot2和ggtree包绘制

图3-17 MAGs 之间的进化树
树状图中距离越近的MAG,进化关系也越亲近;用不同颜色标注出了进化树中的不同微生物门水平分类;右侧热图的颜色与MAG在各样本中的丰度对应。
06-MAG_Circos MAGs基因组Circos图
Circos是由加拿大生物信息学科学家Martin Krzywinski利用Perl语言开发的一款可以用于描述关系型数据和多维数据的基因组圈图可视化软件。2009年Circos发表在Genome research。Circos不仅能将一个物种的整个基因组呈现在一张图片中,还可以给基因组添加丰富的注释信息,如功能注释信息,差异统计信息等等。
利用每个MAG(基于contig)的Prokka蛋白预测信息,功能区注释信息(含正负链、CDS、RNA类型等信息),CAZy数据库注释结果(GH、GT、CE、PL、CBM、AA),以及GC content、GC skew的统计结果绘制Circos圈图。图3-18是其中一个MAG的基因组Circos图,其它MAG请查看结果文件夹。
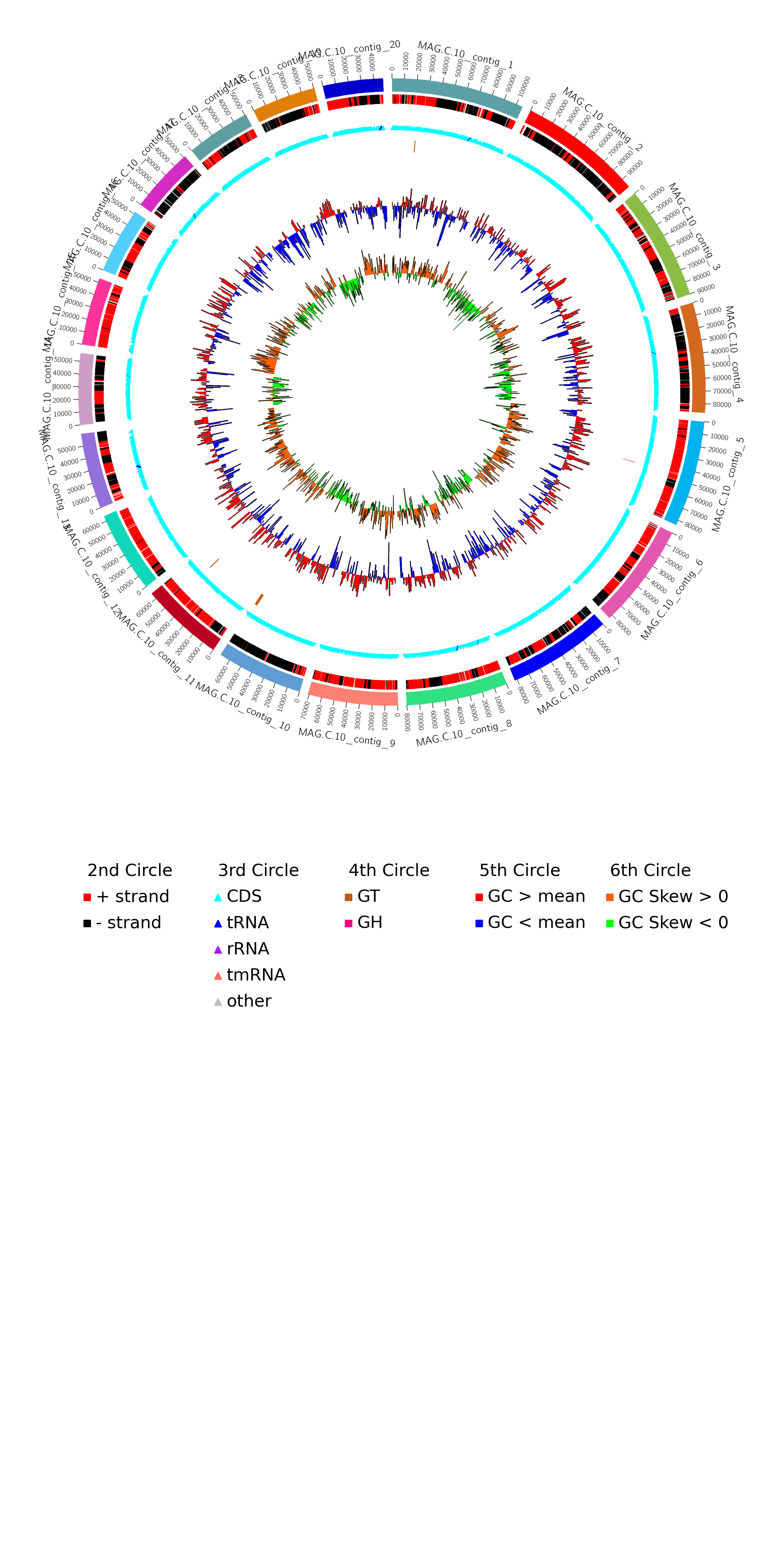
图3-18 MAG基因组圈图
由于图片篇幅限制,图中只展示了长度最长的20条contigs。从外到内,第一圈表示属于该MAG的contigs,长度用刻度表示,单位为bp;第二圈用不同颜色区分contigs上的正链和负链;第三圈用不同颜色的三角标出tRNA, rRNA, tmRNA以及CDS(编码蛋白)的编码区;第四圈标注CDS注释到CAZy数据库的碳水化合物活性酶分类,如果没有注释到任何基因,该圈可能为空;第五圈标注contigs分段(每1kb)GC含量,其中用不同颜色区分大于和小于所有contigs分段GC含量总平均值;第六圈标注contigs 分段(每1kb)GC skew值,大于和小于0用不同颜色区分。
四 常见问题解答
1. 为什么大多数MAGs注释不到种水平?
2. 宏基因组分析得到的优势菌为何无法通过Binning得到其基因组?
五 分析所用软件的版本
| 软件 | 版本 |
|---|---|
| Megahit | 1.2.9 |
| MMseqs2 | 12b7931f517415bc69adf30d62a31701907694b0 |
| BWA | 0.7.17 |
| Samtools | 1.7 |
| MetaBAT2 | 2.15 |
| MaxBin2 | 2.2.7 |
| CONCOCT | 1.1.0 |
| MetaWRAP | 1.3.2 |
| RefineM | 0.1.2 |
| CheckM | 1.1.3 |
| dRep | 2.6.2 |
| SPAdes | 3.13.0 |
| Prokka | 1.14.6 |
| Prodigal | 2.6.3 |
| KofamKOALA | 1.2.0 |
| emapper | 2.0.1 |
| diamond | 0.9.14 |
| PhyloPhlAn3 | 3.0.60 |
| GTDB-Tk | 2.4.0 |
| Trimmomatic | 0.39 |
| Bowtie2 | 2.3.5.1 |
| LEfSe | 1.0.8 |