微科盟宏病毒组分析结题报告
一 概述
病毒宏基因组测序又称宏病毒组(Virome),是在宏基因组学理论的基础上,结合现有的 病毒分子生物学检测技术而兴起的一个新的学科分支。宏病毒组直接以样本中所有病毒的遗 传物质为研究对象,先富集病毒颗粒再获取基因组序列信息后鉴定其中所有的病毒组成及相 对丰度,是一种发现新病毒、病毒感染预警和控制的有力手段,在病毒的起源和进化模式、 遗传多样性和地理分布、以及病毒和宿主的相互关系等研究领域都具有重要意义。宏病毒组 研究可应用于人体或动物血液、组织、粪便等样本,植物组织样本,以及水体、土壤等各种 环境样本,用以分析其中的病毒群落。然而,由于病毒基因组普遍较小,病毒核酸在样本中 的相对含量非常低,宿主的基因组序列干扰严重,且已知病毒的数量和基因组信息有限等问 题,使得宏病毒组研究从样本制备开始到数据分析都存在着一定的困难。
二 项目流程
2.1 测序实验流程
针对不同类型的样品研发出了一套科学有效的病毒样颗粒(Virus-like particle, VLP)分 离和核酸制备技术,可以快速有效地去除宿主细胞、核糖体的干扰,富集样品中的VLP 并制 备宏病毒基因组文库。以组织样品为例(图 2.1),首先需要进行组织破碎和匀浆处理,再经 过低速离心去除组织和细胞的碎片,运用过滤、超速离心等多种病毒学的分离方法对溶液中 的 VLP 进行纯化和浓缩。针对不同类型的样品和所关注病毒类型(如病毒大小、基 因组性 质),在样本前处理流程和方法上会存在一定差异,必要时可进行优化调整。
获得较纯的 VLP 样品后,采用病毒 DNA(dsDNA、 ssDNA)和 RNA(ssRNA、 dsRNA) 共提取的方法同时提取核酸。然后,针对不同病毒基因组类型选择不同的建库方法: dsDNA 基因组可以直接构建 DNA 文库, ssDNA 需要合成双链 DNA 后再进行 DNA 建库, RNA 基因 组则需要经过反转录和二链合成转变成双链 cDNA 后再进行 RNA 建库。如所得 DNA 和/或 RNA 总量及纯度等符合要求则进行后续实验,否则需要根据具体原因进行核酸重新提取,或 对现有核酸进行全基因组扩增、纯化等。
检测合格的 DNA 或经扩增的 DNA 样品采用超声破碎仪进行随机打断,将打断后得到的 短片段 DNA 用于测序文库构建,质检合格的文库将采用 Illumina 平台进行测序,测序读长 PE150,测序结果以 FASTQ(简称为 fq)文件格式存储,其中包含测序序列(Reads)的序列 信息以及其对应的测序质量信息。
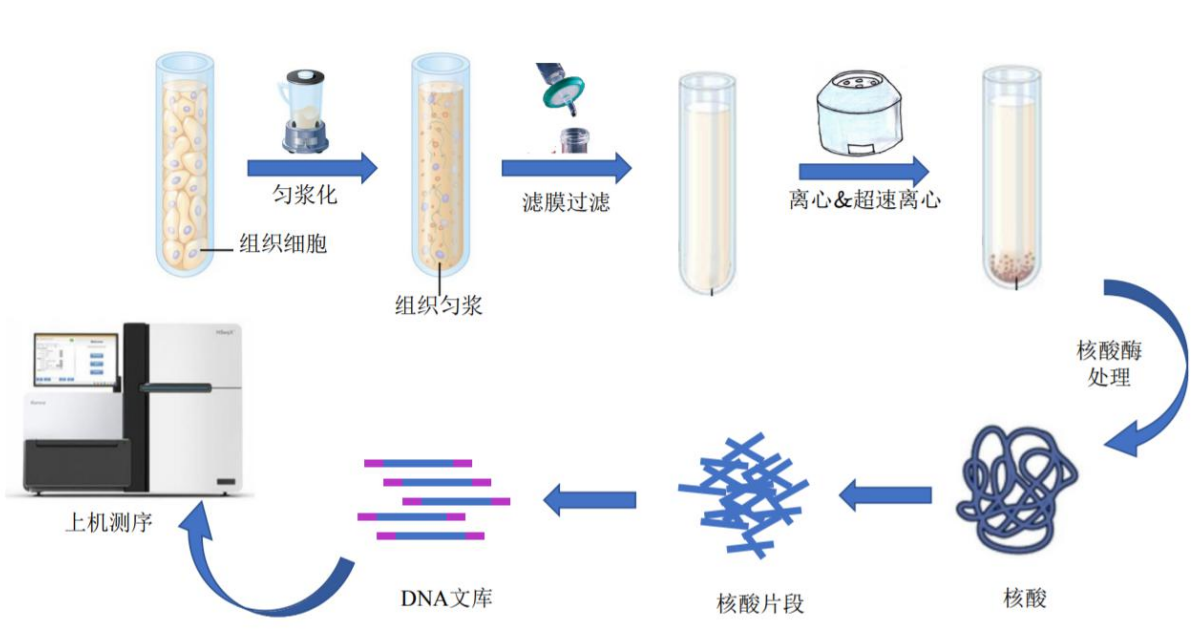
图2-1 宏病毒组组织样品制备流程图
2.2 生信分析流程
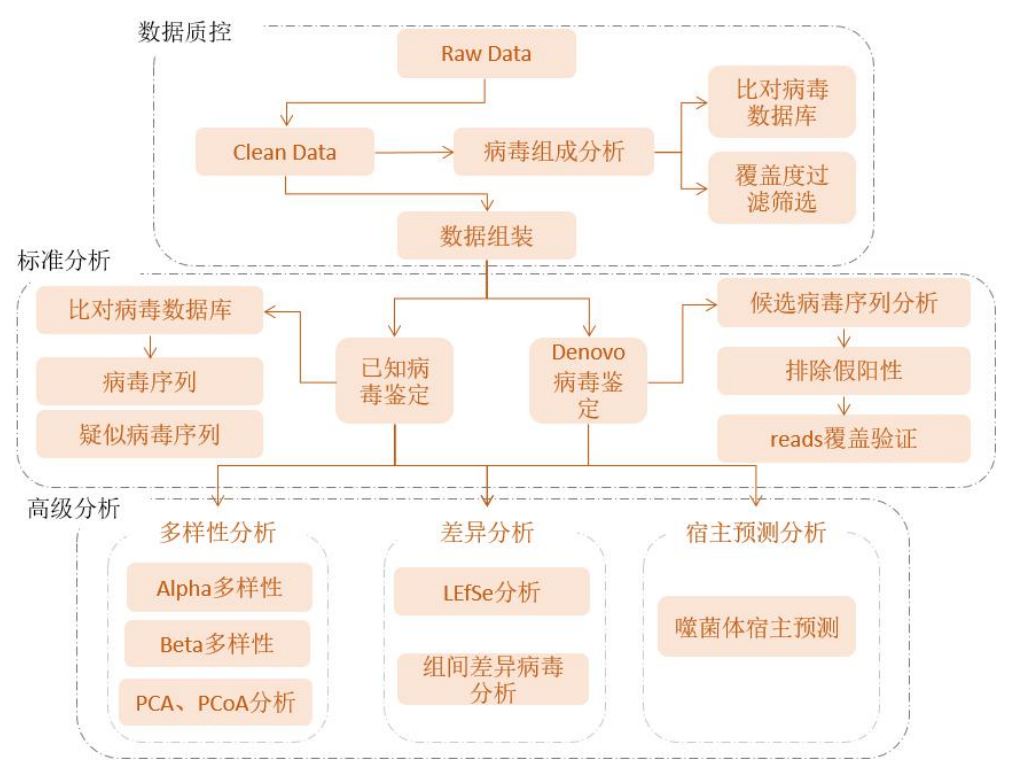
图2-2 生信分析流程图
详细过程见结果解读部分
三 结果文件夹解读
3.1 质控去宿主
01.QC/ ├── after_remove_host.xls [去除宿主序列之后的质量信息统计表格] ├── after_trimmomatic.xls [测序数据质控之后的质量信息统计表格] └── raw.xls [原始数据的质量信息统计表格]
原始序列数据
高通量测序得到的图像经 Base Calling 转化为原始测序序列(Reads),我们称之为 Raw Data 或 Raw Reads,结果以 FASTQ(简写为 fq)文件格式储存,它包含测序序列(Reads) 的序列信息及其对应的测序质量信息。PE 文库的数据结果中,每个样品均有测序两端的 Reads, 并且 Reads 的顺序是严格一致、相互对应的。 FASTQ 文件中每条 read 都由四行构成,文件格 式如下:

图3-1-1 FASTQ格式文件内容示例
其中,第 1 行和第 3 行为 read 名称(后来为了节省储存空间省略掉第 3 行“+”后面的序列 名称),由 Illumina 测序仪产生;第 2 行是 read 的碱基序列,第 4 行是 read 中每个碱基对应 的测序质量分数,由 Base Calling 转化而来,每个字母对应的 ASCII 值减去相应测序质量系 统的 Phred quality score(33 或 64),即为该碱基的测序质量值(Phred 值)。
原始数据的质量信息统计表格如下:
表3-1-1 原始数据的质量信息统计表格(raw.xls)
| sample id | total reads | total bases | total giga bases | q20 bases | q30 bases | q20 rate | q30 rate | read1 mean length | read2 mean length | gc content |
|---|---|---|---|---|---|---|---|---|---|---|
| Sample2 | 10561276 | 1584191400 | 1.58G | 1553247724 | 1494563162 | 0.980467 | 0.943423 | 150 | 150 | 0.658221 |
| Sample3 | 11207766 | 1681164900 | 1.68G | 1647582142 | 1585374075 | 0.980024 | 0.943021 | 150 | 150 | 0.659180 |
| Sample4 | 11925432 | 1788814800 | 1.79G | 1753428415 | 1686857387 | 0.980218 | 0.943003 | 150 | 150 | 0.658728 |
| Sample5 | 10650588 | 1597588200 | 1.6G | 1566620330 | 1507767348 | 0.980616 | 0.943777 | 150 | 150 | 0.660027 |
| Sample6 | 10229314 | 1534397100 | 1.53G | 1506584493 | 1452761052 | 0.981874 | 0.946796 | 150 | 150 | 0.659956 |
| Sample7 | 10291906 | 1543785900 | 1.54G | 1513775570 | 1457045581 | 0.980561 | 0.943813 | 150 | 150 | 0.659168 |
- sample id: 样本ID
- total reads: 样本序列总数
- total bases: 样本碱基总数
- total giga bases: 样本碱基总数(G为单位)
- q20 bases: 质量分高于20(错误率0.01)的碱基总数
- q30 bases: 质量分高于30(错误率0.001)的碱基总数
- q20 rate: 质量分高于20(错误率0.01)的碱基占比(Q20)
- q30 rate: 质量分高于30(错误率0.001)的碱基占比(Q30)
- read1 mean length: read1平均长度
- read2 mean length: read2平均长度
- gc content: GC含量
测序数据质控
在获得每个样品宏基因组测序数据之后,首先需要对测序的数据质量进行评估并对低质 量的数据进行去除,以保证后续分析结果的可信度,以上步骤称之为原始数据的质量控制。 质量控制获得的高质量序列则用于下游的数据分析。质控流程采用软件 Trimmomatic[1] ,具体处理步骤如下:
- 去除带接头(adapter)的 paired reads;
- 当单端测序 read 中含有的低质量( sQ <= 20)碱基数超过该条 read 碱基总数的 20%时,去除此 paired reads;
- 去除 PCR 扩增产生的重复 reads;
- 去除 ployX 序列。
测序数据质控之后的质量信息统计表格如下:
表3-1-2 测序数据质控之后的质量信息统计表(after_trimmomatic.xls)
| sample id | total reads | total bases | total giga bases | q20 bases | q30 bases | q20 rate | q30 rate | read1 mean length | read2 mean length | gc content |
|---|---|---|---|---|---|---|---|---|---|---|
| Sample2 | 10135098 | 1513369435 | 1.51G | 1494931198 | 1445942707 | 0.987816 | 0.955446 | 149 | 149 | 0.658183 |
| Sample3 | 10722060 | 1601165553 | 1.6G | 1581980911 | 1531021743 | 0.988018 | 0.956192 | 149 | 149 | 0.659275 |
| Sample4 | 11432758 | 1707102404 | 1.71G | 1686190254 | 1630918196 | 0.987750 | 0.955372 | 149 | 149 | 0.658735 |
| Sample5 | 10217828 | 1525886195 | 1.53G | 1507545806 | 1458611686 | 0.987981 | 0.955911 | 149 | 149 | 0.660044 |
| Sample6 | 9848174 | 1471079007 | 1.47G | 1454396641 | 1409231449 | 0.988660 | 0.957958 | 149 | 149 | 0.659972 |
| Sample7 | 9875890 | 1474724002 | 1.47G | 1456965970 | 1409697835 | 0.987958 | 0.955906 | 149 | 149 | 0.659143 |
- sample id: 样本ID
- total reads: 样本序列总数
- total bases: 样本碱基总数
- total giga bases: 样本碱基总数(G为单位)
- q20 bases: 质量分高于20(错误率0.01)的碱基总数
- q30 bases: 质量分高于30(错误率0.001)的碱基总数
- q20 rate: 质量分高于20(错误率0.01)的碱基占比(Q20)
- q30 rate: 质量分高于30(错误率0.001)的碱基占比(Q30)
- read1 mean length: read1平均长度
- read2 mean length: read2平均长度
- gc content: GC含量
去除宿主序列
为了避免宿主序列对后续分析的影响,使用 BWA[2](默认参数: mem –k 30)软 件把 clean reads 与宿主(Human)比对,过滤比对长度低于 reads 总长 80%的比对结果,然后 去除相应序列。
去除宿主序列之后的质量信息统计表格如下:
表3-1-3 去除宿主序列之后的质量信息统计表格after_remove_host.xls)
| sample id | total reads | total bases | total giga bases | q20 bases | q30 bases | q20 rate | q30 rate | read1 mean length | read2 mean length | gc content |
|---|---|---|---|---|---|---|---|---|---|---|
| Sample2 | 10134660 | 1513306371 | 1.51G | 1494869225 | 1445883606 | 0.987817 | 0.955447 | 149 | 149 | 0.658186 |
| Sample3 | 10721830 | 1601132608 | 1.6G | 1581948547 | 1530991084 | 0.988018 | 0.956193 | 149 | 149 | 0.659273 |
| Sample4 | 11432500 | 1707065677 | 1.71G | 1686154189 | 1630883972 | 0.987750 | 0.955373 | 149 | 149 | 0.658733 |
| Sample5 | 10217592 | 1525852630 | 1.53G | 1507512674 | 1458579933 | 0.987981 | 0.955911 | 149 | 149 | 0.660043 |
| Sample6 | 9847806 | 1471026617 | 1.47G | 1454345195 | 1409182515 | 0.988660 | 0.957959 | 149 | 149 | 0.659968 |
| Sample7 | 9875658 | 1474690413 | 1.47G | 1456932946 | 1409666533 | 0.987959 | 0.955907 | 149 | 149 | 0.659142 |
- sample id: 样本ID
- total reads: 样本序列总数
- total bases: 样本碱基总数
- total giga bases: 样本碱基总数(G为单位)
- q20 bases: 质量分高于20(错误率0.01)的碱基总数
- q30 bases: 质量分高于30(错误率0.001)的碱基总数
- q20 rate: 质量分高于20(错误率0.01)的碱基占比(Q20)
- q30 rate: 质量分高于30(错误率0.001)的碱基占比(Q30)
- read1 mean length: read1平均长度
- read2 mean length: read2平均长度
- gc content: GC含量
3.2 reads物种分析
02.ReadsClassify/ ├── 01.BasicStatistics [fragments数(read pairs)基础统计图] │ ├── Barplots [百分比堆积柱形图文件夹] │ │ └── [分组方案名] │ │ ├── [物种分类水平] │ │ │ ├── barplot.svg [百分比堆积柱形图] │ │ │ └── table.xls [百分比堆积柱形图对应表格] │ │ ├── Molecule_type [依据各病毒分子类型占比绘制的柱形图] │ │ │ ├── barplot.svg │ │ │ └── table.xls │ │ └──Phages_or_not [依据噬菌体占比绘制的柱形图] │ │ ├── barplot.svg │ │ └── table.xls │ └── Heatmaps [fragments数热图文件夹] │ └── [分组方案名] │ ├── [物种分类水平] │ │ ├── heatmap.svg [fragments数热图] │ │ └── table.xls [fragments数热图对应表格] │ ├── Molecule_type │ │ ├── heatmap.svg │ │ └── table.xls │ └──Phages_or_not │ ├── heatmap.svg │ └── table.xls ├── [分类水平]_count.xls [各物种分类水平比对到NCBI NT数据库序列的fragments数表格] ├── count_mapping.xls [各NCBI NT数据库序列的fragments(read pairs)数,以及分类信息] ├── count_taxonomy.xls [各NCBI NT数据库序列的fragments(read pairs)数,以及物种分类信息] ├── Molecule_type.xls [各病毒分子类型比对到NCBI NT数据库序列的fragments数表格] └── Phages_or_not.xls [噬菌体比对到NCBI NT数据库序列的fragments数表格]
为了快速获得样本中的病毒组成信息,使用 BWA[2](默认参数: mem –k 30)软 件把去除污染后的 clean reads 比对到病毒参考数据(从 NT 数据中分离),过滤比对长度低于 reads 总长 80%的比对结果,统计每个样本每条数据库序列的fragments(read pairs)数。
说明:由于 reads 较短,以及比对软件的局限性,该结果可能会与实际情况出现一些偏差, 这在软件的正常误差范围之内。所以该分析结果仅供参考,以组装后 contigs 的物种注释结果 为准。
根据 NCBI taxonomy 数据库注释信息,统计病毒分类信息:
表 3-2-1 病毒类别的fragments(read pairs)数统计(Phages_or_not.xls)
| Other_virus | Phages | unknown | |
|---|---|---|---|
| Sample5 | 541 | 147228 | 43190 |
| Sample3 | 580 | 154649 | 44354 |
| Sample2 | 515 | 150715 | 42881 |
| Sample6 | 544 | 142008 | 41061 |
| Sample4 | 611 | 169691 | 47725 |
| Sample7 | 493 | 143738 | 41775 |
表 3-2-2 各病毒分子类型的fragments(read pairs)数统计(Molecule_type.xls)
| dsDNA | dsRNA | ssDNA(+) | ssRNA(+) | unknown | |
|---|---|---|---|---|---|
| Sample5 | 111677 | 369 | 14749 | 1 | 64163 |
| Sample3 | 116118 | 404 | 16520 | 3 | 66538 |
| Sample2 | 112180 | 358 | 17304 | 2 | 64267 |
| Sample6 | 107224 | 370 | 14638 | 2 | 61379 |
| Sample4 | 126396 | 440 | 19400 | 3 | 71788 |
| Sample7 | 108803 | 359 | 14529 | 4 | 62311 |

图3-2-1 依据各病毒分子类型fragments数占比绘制的柱形图
如果分类超过20个,则只选取fragments数最高的20个分类绘图

图3-2-2 各样本科分类水平病毒fragments数热图
挑选fragments数最高的20个病毒绘图
3.3 组装
03.Assemble/ ├── contigs [每个样本的contigs文件] │ └── [样本ID].fa [单个样本组装的结果contigs文件] ├── quast [每个样本congigs的quast质量评估结果文件夹] │ └── report.html [每个样本congigs的quast质量评估报告,文件夹中其他文件为报告的数据文件,可忽略] ├── contig_dehost_stat.xls [contig去宿主统计] ├── unique.contigs.fa [所有样本去冗余后的contigs文件] └── reads_used.xls [组装过程reads利用率统计]
使用 Megahit[3] (默认参数: --presets meta-large --min-contig-len 1000)软件对 clean reads 进行组装,使用 BWA[2] 软件把去除污染后的 clean reads 与组装结果比对,计算 reads 利用率(reads_used.xls, 表3-3-1)。同时使用 blast[4] 软件把组装 contigs 与宿主序列比对,去除宿主序列(contig_dehost_stat.xls 统计表, 表3-3-2)。 最后使用 CD-HIT[5](默认参数: -c 0.95 -aS 0.8)软件对所有样本的 contigs 进行去冗余(去冗余后的contig为unique.contigs.fa)。
去宿主序列时,与宿主比对,满足以下一个条件即判定为宿主序列:
- 比对长度>=500bp,比对相似性>=90%;
- 比对长度占 contig 总长的 80%以上,比对相似性>=90%。
表3-3-1 reads 利用率统计(reads_used.xls)
| sample id | total reads | reads mapped to contigs | reads used(%) |
|---|---|---|---|
| Sample2 | 10134660 | 10134522 | 100.0 |
| Sample3 | 10721830 | 10721717 | 100.0 |
| Sample4 | 11432500 | 11432341 | 100.0 |
| Sample5 | 10217592 | 10217486 | 100.0 |
| Sample6 | 9847806 | 9847692 | 100.0 |
| Sample7 | 9875658 | 9875457 | 100.0 |
- sample id: 样本ID
- total reads: clean reads数(去宿主之后的reads数)
- reads mapped to contigs: 比对到该样本contig的reads数
- reads used(%): reads 组装利用率
表3-3-2 宿主contig占比统计表
| sample id | total contigs | host contigs | host percent(%) |
|---|---|---|---|
| Sample2 | 104 | 0 | 0.0 |
| Sample3 | 107 | 0 | 0.0 |
| Sample4 | 100 | 0 | 0.0 |
| Sample5 | 100 | 0 | 0.0 |
| Sample6 | 104 | 0 | 0.0 |
| Sample7 | 98 | 0 | 0.0 |
- sample id: 样本ID
- total contigs: 该样本一共有多少contig
- host contigs: 其中, 属于宿主的contig有多少条
- host percent(%): 宿主的contig占比
对于每个样本最终得到的contigs, 我们使用 QUAST[6] 软件进行质量评估,如图3-3-3。
图3-3-3 每个样本congigs的quast质量评估报告
图中展示了组装得到的contigs 长度分布情况。N50: reads组装后会获得一些不同长度的contigs。 将所有的contigs按照从长到短进行排序,然后把contigs的 长度按照这个顺序依次相加,当相加的长度达到contig总长度的一半时, 最后一个加上的contig长度即为contig N50。 鼠标悬浮在相应字段上,能查看每个字段的详细解释。
3.4 组装物种分析
3.4.1 组装物种注释
04.Taxonomy/
├── 01.Identify
│ ├── 1.suspected
│ │ ├── virus_suspected_blast_tax_filtered.xls [基于参考序列鉴定的suspected病毒分类]
│ │ └── virus_suspected_contig_filtered.fa [基于参考序列鉴定的suspected病毒序列(contig)]
│ ├── 2.confirmed
│ │ ├── virus_confirmed_blast_tax_filtered.xls [基于参考序列鉴定的confirmed病毒分类]
│ │ └── virus_confirmed_contig_filtered.fa [基于参考序列鉴定的confirmed病毒序列]
│ ├── 3.denovo
│ │ ├── virus_denovo_blast_tax_filtered.xls [Denovo方法鉴定的病毒分类]
│ │ └── virus_denovo_contig_filtered.fa [Denovo方法鉴定的病毒序列]
│ ├── 4.combined
│ │ ├── combined_blast_tax.xls [结合Denovo方法和基于参考序列方法鉴定的病毒分类]
│ │ ├── combined_contig.fa [结合Denovo方法和基于参考序列方法鉴定的病毒序列]
│ │ └── venn.svg [suspected, confirmed, denovo鉴定的病毒序列集合venn图]
│ └── quast
│ └── report.html [各途径获得的病毒 contig序列质量评估报告,文件夹中其他文件为报告的数据文件,可忽略]
├── 02.ContigCount [各类型病毒的contig计数分析文件夹]
│ ├── Molecule_type.xls [各病毒分子类型contig计数表]
│ ├── Phages_or_not.xls [噬菌体contig计数表]
│ └── [病毒分类水平]_count.xls [各病毒分类水平(界门纲目科属种)contig计数表]
└── 03.Abundance [病毒丰度分析结果文件夹]
├── abundance_taxonomy.xls [病毒contig TPM丰度表]
├── Molecule_type.xls [各病毒分子类型TPM丰度表]
├── Phages_or_not.xls [噬菌体TPM丰度表]
└── [病毒分类水平]_abundance.xls [各病毒分类水平(界门纲目科属种)TPM丰度表]
基于参考序列的鉴定
使用 blast[4]软件, 将获得的 unique.contigs(去冗余后的contig) 与病毒数据库(从 NT 数据库中分离)比对。
比对筛选标准如下:
- 比对相似性>=80%,比对长度>=500bp, e(evalue 简称)<=1e-5,可信度较高,定义为 confirmed(virus_confirmed_contig_filtered.fa);
- 不满足 A 中的条件,但是比对长度>=100bp, e<=1e-5,可信度低,则定义为 suspected,需要深入分析验证(virus_suspected_contig_filtered.fa);
- 根据 NCBI taxonomy 注释信息(2022/6/22 更新),分别注释病毒分子类型,噬菌体,和病毒分类(*_blast_tax_filtered.xls)。
*_blast_tax_filtered.xls表格的各列解释如下:
- qseqid: 查询序列ID(contig ID)
- sseqid: 比对到的数据库序列ID
- identity: identity=比对长度/查询序列长度与数据库序列长度的较小值,可理解为相似度
- length: 比对长度
- mismatch: 错配碱基数
- gapopen: 比对区域的gap数目
- qstart: 比对区域在查询序列(qseqid)上的起始位点
- qend: 比对区域在查询序列(qseqid)上的终止位点
- sstart: 比对区域在数据库序列(sseqid)上的起始位点
- send: 比对区域在数据库序列(sseqid)上的终止位点
- evalue: 随机序列中,存在比该数据库序列更好的匹配序列的概率,越低该比对结果越可靠
- bitscore: 比对得分,越高越可靠
- Phages_or_not: 噬菌体
- Molecule_type: 病毒分子类型
- Taxonomy: 病毒分类
Denovo 病毒序列鉴定
基于参考数据的比对,存在两个缺陷:一是只能鉴定已知病毒;二是结果中存在假阳性。 为了降低假阳性以及鉴定未知病毒,接下来综合利用多个数据库,使用新的方法来进一步鉴 定病毒序列。
主要参考使用如下几篇文献,对前述获得的 unique.contigs 中的病毒序列进行鉴定:
- Uncovering Earth’s virome(Nature, 2016),使用 hidden Markov models(HMMs) 构建病毒的蛋白家族数据库(Viral protein family models, VPFs)
- Redefining the invertebrate RNA virosphere(Nature, 2016)
- VirusSeeker, a computational pipeline for virus discovery and virome composition analysis(Virology, 2017)
- Profile hidden Markov models for the detection of viruses within metagenomic sequence data(PLoS ONE, 2014),使用 hidden Markov models(HMMs)构建病毒的蛋白家族数据库 vFam。
分析过程如下:
Step1:寻找候选病毒序列:将 contigs 与多个数据库比对,只需满足如下三个条件中一 个即可:
- 使用 blastn[4]与分离自 NT 的病毒数据库(Virus-NT,包含噬菌体)比对, 筛选 e <= 1e-5 的比对结果;
- 使用 blastx[4] 与分离自 NR 的病毒数据库(Virus-NR,包含噬菌体)比对, 筛选 e <= 1e-3 的比对结果;
- 使用 MetaGeneMark[7] 预测基因,然后使用 hmmsearch[8]软件,将蛋 白序列与 HMM 数据库比对(VPFs [9]和 vFam[10]),筛选 e <= 1e-5 的比对结果。
Step2:排除假阳性
- 将上述获得候选病毒序列与 NT 数据库 blast[4]比对,筛选 e <= 1e-10 的比对结果;
- 将前一步未比对上的序列与 NR 数据库使用 diamond[11]比对,筛选 e <= 1e-3 的比对结果;
- 使用 NCBI Taxonomy 数据对前述比对结果进行注释,如果在前 50 个比对结果中,有 20%以上的比对结果为非病毒序列(注释结果为 Eukaryota、 Bacteria、 Archaea),则该序列被 认为是非病毒序列,其余序列被认为是病毒序列(virus_denovo_contig_filtered.fa);
- 根据病毒 contigs 与 Virus-NT 的 best hit(evalue 最小) 比对结果(length>=100 bp, e<=1e-5),对病毒 contigs 进行注释(virus_denovo_blast_tax_filtered.xls)。
结合基于参考序列(Confirmed)和Denovo的方法
比较上述两种方法获得的有分类病毒或未知病毒 contigs 序列集合的差异,绘制韦恩图(图3-4-1),保留Denovo 和 Confirmed 中的病毒序列,作为最终的病毒 鉴定结果(combined_contig.fa)。

图3-4-1 不同方法获得的 病毒 contigs 数量比较
- denovo:denovo方法获得的病毒序列集合;
- confirmed: confirmed 病毒序列集合;
- suspected: suspected 病毒序列集合。
各途径获得的病毒 contig序列质量评估
对于各途径获得的病毒 contig序列, 我们使用 QUAST[6] 软件进行质量评估,如图3-4-2。
图3-4-2 每个样本congigs的quast质量评估报告
图中展示了各途径获得的病毒 contig序列 长度分布情况。N50: 将所有的contigs按照从长到短进行排序,然后把contigs的 长度按照这个顺序依次相加,当相加的长度达到contig总长度的一半时, 最后一个加上的contig长度即为contig N50。 鼠标悬浮在相应字段上,能查看每个字段的详细解释。
各类型病毒的contig计数统计
对于每种途径(基于参考序列的方法,Denovo方法, 以及结合基于参考序列和Denovo的方法)获得的病毒contig序列, 我们统计了各个病毒分子类型(dsDNA, ssRNA等,Molecule_type.xls),噬菌体(Phages_or_not.xls), 各个分类水平([病毒分类水平]_count.xls)的每个分类的contig计数(04.Taxonomy/02.ContigCount),用于后续可视化分析。 由于Denovo方法鉴定的未知病毒没有分类信息,故不参与contig计数统计
病毒丰度
对于结合基于参考序列(Confirmed)和Denovo的方法获得的病毒contig序列, 我们通过如下步骤计算病毒contig的丰度, 并依据contig分类信息,统计每种类型病毒的丰度,用于后续可视化分析。
- 使用 BWA[2](默认参数: mem -k 30)软件,将去除污染后的 clean reads 分 别与病毒 contigs 比对,过滤比对长度低于 reads 总长 80%的比对结果,统计病毒 fragments (read pairs)数;
- 计算每条病毒 contig 的 TPM (云流程可选择FPKM, fragment count)值(abundance_taxonomy.xls), FPKM和TPM的计算请参照 文献中的公式
- 依据contig分类信息,统计每种类型病毒的丰度。(04.Taxonomy/03.Abundance)
3.4.2 contig计数可视化
02.ContigCount/ [各类型病毒的contig计数统计文件夹] ├── bar [contig计数柱形图文件夹] │ ├── Molecule_type [依据各病毒分子类型contig计数绘制的柱形图文件夹] │ │ ├── combined_bar.svg [结合Denovo方法和基于参考序列方法鉴定的病毒contig计数柱形图] │ │ ├── virus_confirmed_bar.svg [基于参考序列鉴定的confirmed病毒contig计数柱形图] │ │ ├── virus_denovo_bar.svg [Denovo方法鉴定的病毒contig计数柱形图] │ │ └── virus_suspected_bar.svg [基于参考序列鉴定的suspected病毒contig计数柱形图] │ ├── Phages_or_not [依据噬菌体的contig计数绘制的柱形图文件夹] │ │ ├── combined_bar.svg │ │ ├── virus_confirmed_bar.svg │ │ ├── virus_denovo_bar.svg │ │ └── virus_suspected_bar.svg │ └── [病毒分类水平] [依据各病毒分类水平(界门纲目科属种)contig计数绘制的柱形图文件夹] │ ├── combined_bar.svg │ ├── virus_confirmed_bar.svg │ ├── virus_denovo_bar.svg │ └── virus_suspected_bar.svg ├── pie [contig计数占比饼图文件夹] │ ├── Molecule_type [依据各病毒分子类型contig计数占比绘制的饼图文件夹] │ │ ├── combined_pie.svg [结合Denovo方法和基于参考序列方法鉴定的病毒contig计数占比饼图] │ │ ├── virus_confirmed_pie.svg [基于参考序列鉴定的confirmed病毒contig计数占比饼图] │ │ ├── virus_denovo_pie.svg [Denovo方法鉴定的病毒contig计数占比饼图] │ │ └── virus_suspected_pie.svg [基于参考序列鉴定的suspected病毒contig计数占比饼图] │ ├── Phages_or_not [依据噬菌体的contig计数占比绘制的饼图文件夹] │ │ ├── combined_pie.svg │ │ ├── virus_confirmed_pie.svg │ │ ├── virus_denovo_pie.svg │ │ └── virus_suspected_pie.svg │ └── [病毒分类水平] [依据各病毒分类水平(界门纲目科属种)contig计数占比绘制的饼图文件夹] │ ├── combined_pie.svg │ ├── virus_confirmed_pie.svg │ ├── virus_denovo_pie.svg │ └── virus_suspected_pie.svg ├── Molecule_type.xls [参照3.4.1小节解读] ├── Phages_or_not.xls [参照3.4.1小节解读] └── [病毒分类水平]_count.xls [参照3.4.1小节解读] 注:同名文件解释相同,不赘述
可视化部分的解读,我们只挑选了部分有代表性的图来说明,更多详细内容请直接看结果文件夹。
柱形图
柱形图用于展示每个病毒类别的contig数量。

图3-4-3 结合Denovo方法和基于参考序列方法鉴定的科水平病毒contig数量柱形图
若科的数目超过20个,只展示contig数量最高的前20个。
饼图
饼图用于展示每个病毒类别的contig数量占比。若类别数目超过20个,只展示contig数量占比最高的前20个

图3-4-4 结合Denovo方法和基于参考序列方法鉴定的噬菌体contig数量占比饼图

图3-4-5 结合Denovo方法和基于参考序列方法鉴定的各病毒分子类别contig数量占比饼图
3.4.3 病毒丰度可视化
03.Abundance/ ├── 01.BasicStatistics [基础统计文件夹] │ ├── Barplots [百分比堆积柱形图文件夹] │ │ └── [分组方案名] │ │ └── [分类水平或方式] │ │ ├── barplot.svg [百分比堆积柱形图] │ │ └── table.xls [百分比堆积柱形图数据] │ └── Heatmaps [丰度热图文件夹] │ └── [分组方案名] │ └── [分类水平或方式] │ ├── heatmap.svg [丰度热图] │ └── table.xls [丰度热图数据] ├── 02.GroupComparison [差异比较文件夹] │ ├── ANOVA_Duncan [ANOVA和Duncan检验结果文件夹] │ │ └── [分组方案名] │ │ └── [分类水平] │ │ ├── [分组方案名]_all_feature_anova_results.xls [ANOVA检验结果] │ │ ├── [分组方案名]_all_significant_feature_duncan_results.xls [Duncan检验结果, ANOVA无显著可缺失] │ │ ├── [分组方案名]_all_significant_feature_barplot_of_duncan.svg [Duncan检验结果图, ANOVA无显著可缺失] │ │ └── final_data_for_anova.xls [检验数据(原始丰度log转化,标准化之后)] │ ├── DunnTest [Kruskal-Wallis和Dunn’s Test检验结果文件夹] │ │ └── [分组方案名] │ │ └── [分类水平] │ │ └── DunnTest.xls [Kruskal-Wallis和Dunn’s Test检验结果表格] │ └── LEfSe [LEfSe分析结果文件夹] │ └── [分组方案名] │ └── [分类水平] │ ├── data_for_lefse.xls [LEfSe分析输入文件] │ ├── lefse.cladogram.svg [LEfSe分析结果cladogram图] │ ├── lefse.LDA.xls [LEfSe分析结果表格] │ ├── lefse.normalized.txt [LEfSe标准化后的数据,中间文件] │ └── lefse.svg [LEfSe分析结果LDA图] ├── 03.Correlation [相关性分析结果文件夹] │ ├── CorrelationHeatmap [相关性热图结果文件夹] │ │ └── [分类水平] │ │ ├── Correlation_heatmap.svg [相关性热图] │ │ ├── p_value_matrix.xls [相关系数p值矩阵] │ │ └── spearman_rank_correlation_matrix.xls [spearman相关系数矩阵] │ └── RDA [CCA/RDA结果文件夹] │ └── [分组方案名] │ └── [分类水平] │ ├── [分组方案名]_RDA.envfit.xls [RDA各环境因子坐标,解释方差,p值] │ ├── [分组方案名]_RDA.Factors.PERMANOVA.xls [所有环境因子对病毒群落变异的解释方差的p值计算结果] │ ├── [分组方案名]_RDA_features_location_plot.svg [RDA病毒点图] │ ├── [分组方案名]_RDA.features.xls [RDA病毒点坐标] │ ├── [分组方案名]_RDA.Group.PERMANOVA.xls [分组方式对病毒群落变异的解释方差的p值计算结果] │ ├── [分组方案名]_RDA_sample_location_plot.svg [RDA样本点图] │ ├── [分组方案名]_RDA_sample_location_plot_with_labels.svg [RDA样本点图(带样本名)] │ ├── [分组方案名]_RDA.sample.xls [RDA样本点坐标] │ └── DCA_output.xls [DCA分析各排序轴值] ├── 04.Diversity [多样性分析结果文件夹] │ ├── Alpha [α多样性分析结果文件夹] │ │ └── [分组方案名] │ │ ├── 1-AlphaDiversitySummary │ │ │ ├── alpha-summary.xls [α多样性指数表格] │ │ │ ├── [分组方案名]_alpha_diversity_shannon_entropy.boxplot.svg [shannon指数箱线图] │ │ │ └── [分组方案名]_alpha_diversity_simpson.boxplot.svg [simpson指数箱线图] │ │ ├── 2-AlphaRarefaction [α多样性稀释性曲线] │ │ │ ├── alpha-rarefaction-ggplot2 [ggplot2包画的稀释性曲线拟合曲线文件夹] │ │ │ │ ├── group_mean_observed_features_rarefaction_curve.svg [observed features(contig数)分组均值稀释性曲线拟合曲线] │ │ │ │ ├── group_mean_shannon_rarefaction_curve.svg [shannon指数分组均值稀释性曲线拟合曲线] │ │ │ │ ├── observed_features_rarefaction_curve.svg [observed features(contig数)稀释性曲线拟合曲线] │ │ │ │ └── shannon_rarefaction_curve.svg [shannon指数稀释性曲线拟合曲线] │ │ │ └── alpha-rarefaction-Qiime2 [qiime2画的稀释性曲线文件夹] │ │ │ └── Summary_请点此文件查看.html [qiime2画的稀释性曲线, 文件夹内其它文件为网页数据,可忽略] │ │ └── 3-SignificanceAnalysis [α多样性组间差异比较文件夹] │ │ ├── 1-Wilcox_Test │ │ │ ├── alpha_Category_wilcox_compare_results.xls [Wilcox Test结果表] │ │ │ ├── shannon_entropy_Category_wilcox_compare_boxplot.svg [shannon指数Wilcox Test结果图] │ │ │ └── simpson_Category_wilcox_compare_boxplot.svg [simpson指数Wilcox Test结果图] │ │ └── 2-Kruskal_Wallis │ │ ├── shannon-group-significance │ │ │ └── Summary_请点此文件查看.html [shannon指数Kruskal Wallis检验报告, 文件夹内其它文件为网页数据,可忽略] │ │ └── simpson-group-significance │ │ └── Summary_请点此文件查看.html [simpson指数Kruskal Wallis检验报告, 文件夹内其它文件为网页数据,可忽略] │ └── Beta [β多样性分析结果文件夹] │ ├── NMDS │ │ └── [分组方案名] │ │ ├── bray_curtis_distance_matrix.xls [Bray-Curtis距离矩阵] │ │ ├── nmds_points_ordinates.xls [NMDS分析样本点坐标] │ │ └── NMDS.svg [NMDS分析结果图] │ ├── PCA │ │ └── [分组方案名] │ │ ├── PCA_3D.svg [三维PCA图] │ │ ├── pca_points_ordinates.xls [PCA分析样本点坐标] │ │ └── PCA.svg [二维PCA图] │ └── PCoA │ └── [分组方案名] │ ├── bray_curtis_distance_matrix.xls [Bray-Curtis距离矩阵] │ ├── PCoA_3D.svg [三维PCoA图] │ ├── pcoa_points_ordinates.xls [PCoA分析样本点坐标] │ └── PCoA.svg [二维PCoA图] ├── abundance_taxonomy.xls [参照3.4.1小节解读] ├── Molecule_type.xls [参照3.4.1小节解读] ├── Phages_or_not.xls [参照3.4.1小节解读] └── [病毒分类水平]_abundance.xls [参照3.4.1小节解读]
基础统计
百分比堆积柱形图
百分比堆积柱形图用于展示各样本中,每个病毒类别的丰度占比。

图3-4-6 科水平病毒百分比堆积柱形图
当病毒类别超过20个时,只展示占比最高的前20个
丰度热图
丰度热图用于展示各样本中,每个病毒类别的丰度。为了方便比较, 热图中展示的丰度是标准化后的Z-Score,即丰度减去平均值,除以标准差。

图3-4-7 科水平病毒丰度热图
当病毒类别超过20个时,只展示丰度最高的前20个
差异比较
LEfSe
LEfSe[13] (Linear discriminant analysis Effect Size,线性判别分析)即 LDA Effect Size 分析, 是一种发现和解释高纬度数据生物标识(分类单元、通路、基因)的分析工具,可以实现两 个或者多个分组之间的比较,同时也可进行分组内部亚组之间的比较分析。该分析首先使用非参数 Kruskal-Wallis 秩和 检测不同分组间丰度差异显著的病毒 ,然后使用 Wilcoxon 秩和检验上一步的差异病毒 在不同组间子分组中的差异一致性,最后采用线性回归分析(LDA)来估算每个病毒 的丰度对差异效果影响的大小。 LEfSe寻找每一个分组的特征病毒(默认为LDA>2的病毒), 也就是相对于其他分组,在这个组中丰度较高的病毒。 需要注意的是,LEfSe内部会对丰度进行标准化(主要计算每个样本参与分析病毒的丰度比例), 特征病毒原始丰度(TPM)少数情况下不是最高的。

图3-4-8 种水平病毒LEfSe分析LDA柱形图
每一横向柱形体代表一个物种,柱形体的长度对应LDA值,LDA值越高则差异越大。 柱形的颜色对应该物种是哪个分组的特征病毒(在对应分组中的丰度相对较高)。

图3-4-9 种水平病毒LEfSe分析cladogram图
cladogram图由内到外,分别对应界门纲目科属种不同的分类层级,层级间连线代表所属关系。 每个圆圈节点代表一个物种,节点为黄色代表在分组间差异不显著, 不为黄色则代表该物种是对应颜色分组的特征病毒(在该分组丰度显著较高)。 有颜色的扇形区域标注了特征病毒的下属分类区间。
lefse.LDA.xls表格文件从左到右各列解释为:
- feature标识
- 所有样本标准化(算比例)以及log转化后的丰度均值
- 特征feature对应分组,如果为空,表示该feature不是特征feature
- LDA值
- Kruskal-Wallis 秩和检验p值
Kruskal-Wallis和Dunn’s Test检验
使用R语言dunn.test包[14]进行Kruskal-Wallis和Dunn’s Test检验。 Kruskal-Wallis和Dunn’s Test都是基于秩次的非参数检验,对数据分布没有要求。 Kruskal-Wallis 秩和检验通常只能检验一个分组方案所有分组的整体差异, p<0.05只能推断至少有两个分组不一样,如果分组个数大于2, 那具体哪两个分组不一样是无法推断的。这时候我们需要结合Dunn’s Test来进行 多重比较,进而推断具体哪两个分组不一样。
Dunn’s Test的结果表格(DunnTest.xls)各列解释如下:
- Feature: feature标识
- KW_pvalue: Kruskal-Wallis秩和检验p值
- DunnTest_comparison: 分组对
- DunnTest_Z: 分组对Dunn’s Test检验统计量
- DunnTest_PValue: 分组对Dunn’s Test检验p值
- DunnTest_PValueBonferroniAdjusted: 分组对Dunn’s Test检验Bonferroni校正后的p值
ANOVA和Duncan检验
使用R语言ggpubr包[15]进行ANOVA检验, 使用R语言agricolae包[16]进行Duncan检验。 ANOVA和Duncan的关系类似Kruskal-Wallis和Dunn’s Test的关系。区别是,ANOVA和Duncan是有参的检验方法。 要求数据服从正态分布。我们在做该分析之前,筛除检出率(丰度大于0的频率)小于0.2的 features, 并对丰度进行log转化以及标准化(减去平均值,除以标准差),使得数据尽量接近正态分布。
*_all_feature_anova_results.xls为ANOVA检验结果,各列解释如下:
- variable: features标识
- .y.: 无意义
- p: p值
- p.adj: holm方法校正后的p值
- p.format: 保留三位小数的p值
- p.signif: 显著性
- method: 检验使用的方法
*_all_significant_feature_duncan_results.xls为Duncan检验结果,各列解释如下:
- Feature: features标识
- Group: 分组名
- Mean: 分组均值
- SE: 分组标准误
- Duncan significance: 用字母表示的显著性,同一个feature,字母相同的分组差异不显著,不同则显著
图3-4-10 ANOVA和Duncan检验得到的所有显著差异的病毒
横坐标为病毒名字;对于每个病毒,用不同颜色表示不同的分组,两个分组上方如果有相同的字母,则代表差异不显著,否则差异显著。
相关性分析
相关性热图
如果提供了环境因子的数据,比如 pH值、温度值、临床因子等,可以进行相关性热图分析
相关性热图的绘制主要运用了R语言pheatmap包[17]。 相关性热图可以用于分析环境因子或其他临床表型数据与病毒群落或物种之间是否显著相关, 然后计算环境因子与病毒物种间的Spearman相关系数,并用热图展示。 该分析可以挑选出与某种环境因子或疾病显著相关的物种。

图3-4-11 病毒与表型之间的相关性热图
X轴上为环境因子,Y轴为物种。通过计算获得R值(秩相关)和p值。 R值在图中以不同颜色展示,p值若小于0.05则用 * 号标出, 右侧图例是不同R值的颜色区间,同时,左边的色条标注了物种所属门分类。 * 0.01≤ p <0.05,** 0.001≤p < 0.01,*** p < 0.001。 先依据显著的环境因子个数排名(越多越靠前),再依据p值平均值排名(越低越靠前), 该图只展示排名前20个病毒,如果没有显著相关的病毒,则无法作图。
RDA/CCA分析
CCA/RDA的分析主要依赖R语言vegan包[18], 以及用ggplot2[19]进行可视化。 CCA/RDA(DCA判断用哪一种分析)分析是基于对应分析发展的一种排序方法, 将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析。 RDA是基于线性模型,CCA是基于单峰模型(图7-1)。本报告先进行DCA分析,看最大轴的值是否大于4,如果大于4.0, 就选CCA,否则选RDA。该分析主要用来反映病毒群落与环境因子之间的关系, 可以检测环境因子、样品、病毒群落(或功能)三者之间的关系或者两两之间的关系, 可得到影响样品分布的重要环境驱动因子。该分析给出的所有p值都是反映解释变量 (连续的环境因子,或者分组方案)对病毒群落变异的解释程度是否显著 (简单的说就是解释变量对病毒群落是否有影响,影响是否显著),所有p值都是用R语言vegan包里的置换检验得出的 (permutation test),*_features_location_plot图中的p值 反映了所有连续的数值变量(环境因子)对病毒差异的解释程度(总的p值), 表格*_RDA.envfit中的p值反映了每个环境因子对病毒差异的解释程度, *RDA_sample_location_plot图中的p值反映了分组对病毒差异的解释程度,p<0.05, 解释方差显著。

图3-4-12 种水平病毒CCA/RDA排序图
在CCA/RDA物种排序图内,环境因子用箭头表示, 箭头连线的长度代表某个环境因子与群落分布和种类分布间相关程度的大小(解释方差的大小), 箭头越长,说明相关性越大,反之越小。箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小, 夹角越小,相关性越高;反之越低。环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系, 钝角时呈负相关关系。每个点代表一个物种,点越大,对应物种丰度越高, 灰色点代表丰度较低的物种,未在图中注释物种名称,将物种投影到各个环境因子, 对应的值即为该物种倾向于存在的环境(喜欢的环境)。或者说,将物种点与原点连线, 物种间,物种与环境因子间,环境因子间的夹角的余弦值近似于相关系数,至于有多近似, 就要看RDA1/CCA1和RDA2/CCA2两个坐标轴的解释方差百分比有多大,越大越近似。 对于样本排序图,样本点之间的距离近似于病毒群落结构差异程度, 样本点投影到环境因子对应的值近似于该样本真实的环境因子值。

图3-4-13 CCA/RDA样本排序图
环境因子用箭头表示, 箭头连线的长度代表某个环境因子与群落分布和种类分布间相关程度的大小(解释方差的大小), 箭头越长,说明相关性越大,反之越小。箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小, 夹角越小,相关性越高;反之越低。环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系, 钝角时呈负相关关系。样本点之间的距离近似于病毒群落结构差异程度, 样本点投影到环境因子对应的值近似于该样本真实的环境因子值。
*_RDA.envfit.xls表格各列解释如下:
- 环境因子
- RDA1: 表征环境因子的方向,与横轴正方向夹角的余弦值
- RDA2: 表征环境因子的方向,与横轴正方向夹角的正弦值
- r2: 环境因子对病毒群落变异的解释方差
- p value: 置换检验计算的解释方差的p值,小于0.05,可推断该环境因子对病毒群落影响显著
*_RDA.*.PERMANOVA.xls表格为所有环境因子(或分组方式)对病毒群落结构变异的解释方差的p值计算结果, 用的是PERMANOVA置换检验的方法,各列解释如下:
- 方差组分(模型部分,剩余部分)
- Df: 自由度
- Variance: 方差剖分
- F: PERMANOVA检验统计量
- Pr(>F): PERMANOVA检验p值
多样性分析
α多样性分析
α 多样性(α diversity)是对单个样品中物种多样性的分析,包含物种组成的丰 富度(species richness)和均匀度(species evenness)两个因素。 shannon (shannon_entropy)和simpson 指数可综合考虑群 落中物种的丰富度和均匀度。群落中有越多的物种(丰富度越高),各物种具有越大的均匀度, 则群落具有越大的α 多样性, shannon和simpson 指数也越高。 α 多样性分析通常用于扩增子测序分析,在宏病毒组的α 多样性分析中,我们使用unique.contig来近似 ASV的概念(多少有些偏差,结果只能作为参考)。 本分析使用qiime2[20]软件, 基于样品病毒contig序列的 TPM 值(abundance_taxonomy.xls,云流程可调整)计算shannon和simpson 指数。

图3-4-14 shannon指数的箱型图
不同颜色代表不同的分组。
Alpha多样性稀释曲线(Rarefaction Curve)是从样品中随机抽取一定测序量的数据(序列数标准化为TPM,测序量为一百万), 统计它们所属物种数目(unique.contig数目)或多样性指数,以测序数据量与物种多样性来构建的曲线, 以用来说明样品的测序数据量是否合理,并间接反映样品中物种的丰富程度。 曲线的平缓程度反映了测序深度对于观测样本多样性的影响大小,曲线越平缓, 表明测序结果已足够反映当前样本所包含的多样性,继续增加测序深度已无法检测到大量的尚未发现的新物种; 反之,则表明多样性尚未接近饱和,继续增加测序深度将有助于观测到更多的新物种。
图3-4-15 各α多样性指数的稀释性曲线
通过选择下拉框选择不同的α多样性指数或者分组情况(网页上半部分图), 图中X轴代表抽取的序列测序量,Y轴代表相应的α多样性指数值。 网页下半部分图代表在相应抽取测序量时,各分组中仍包含样品数量, 用于查看在一定抽取测序量之下稀释曲线的分组分析是否仍然包括该分组中全部的样本。
图3-4-16是用R语言ggplot2[19] 包画的α多样性指数稀释性曲线的拟合曲线(公式为y ~ log(x)), 这个曲线比较光滑,但也和真实数据存在一定偏差。

图3-4-16 shannon指数稀释性曲线拟合曲线
在得到整体的α多样性指数之后,接下来结合分组信息来用Kruskal Wallis方法 比较在各个样品分组之间α多样性指数是否有显著性差异。 除了使用qiime2推荐的Kruskal Wallis方法,分析还包括使用Wilcox Test 去精确比较了各个组间的显著性差异并做图,图3-4-17显示了shannon指数的组间多重比较结果。

图3-4-17 shannon指数的组间Wilcox多重比较
X轴表示分组名称,Y轴表示α多样性指数。*,**,***分别代表p<0.05, p<0.01, p<0.001。
β多样性分析
β 多样性(β Diversity)即样品间的生物多样性的比较,是对不同样品间的微生物 群落构成进行比较。样本间物种的丰度分布差异程度可通过统计学中的距离进行量化分析, 使用统计算法计算两两样本间距离,获得距离矩阵,可用于后续进一步的 β 多样性分析 和可视化统计分析。 Bray-Curtis 距离是系统聚类法中使用最普遍的一个距离指标,基于物种 的丰度信息计算,是生态学上反映群落之间结构差异性常用的指标之一。 使用R语言vegan[18]包,根据样本病毒contig序列 TPM 值(abundance_taxonomy.xls,云流程可调整)计算 Bray-Curtis 距离,来评估不同样品间的病毒群落结构差异。
PCA(Principal Component Analysis,主成分分析)是一种传统的数据降维方法,常用于 简化数据集。它运用方差分解,寻找造成样本间差异的主成分(特征值)及其贡献率,选取 两个或三个贡献率最大的主成分进行作图,将多组数据的差异反映在二维或三维坐标图上。

图3-4-18 PCA 分析结果
- 一个点代表一个样本,点与点空间距离近似物种组成结构的差异程度(欧式距离);
- 主成分 1(PC1)和主成分 2(PC2)是造成样品间差异的第一和第二大成分,百分比表示两个主成分对样品差异(方差)的 贡献率。
PCoA(Principal Co-ordinates Analysis,主坐标分析)与 PCA 类似,都是通过降维找出影 响样本群落组成差异的潜在主成分,二者的主要区别在于 PCA 只基于欧式距离, 而 PCoA 可以基于多种距离矩阵,PCA可以理解为一种特殊的PCoA(基于欧式距离的PCoA就是PCA)。 我们使用R语言ape[21]包, 基于 Bray-Curtis 距离来进行 PCoA 分析, 同时使用R语言vegan[18]包, 基于 Bray-Curtis 距离来进行PERMANOVA分析(即adonis), 以检验分组间病毒群落结构的差异是否显著。

图3-4-19 PCoA 分析结果
- 一个点代表一个样本,点与点空间距离近似物种组成结构的差异程度(Bray-Curtis 距离);
- 两个坐标轴百分比表示两个主成分对样品微生物群落差异(方差)的贡献率。
- 图中PERMANOVA分析的p值用于检验分组间病毒群落结构的差异是否显著
无度量多维标定法(NMDS)统计是一种适用于生态学研究的排序方法,类似于PCoA,通过样本微生物群落结构, 或者基因的组成,可以看出组间差异,组内差异等。NMDS包括一类排序方法, 其设计目的是为了克服以前的排序方法(即线性模型,包括PCoA)的缺点,NMDS的模型是非线性(只考虑距离的秩次,而不在乎距离的大小差异)的, 能更好地反映生态学数据的非线性结构。非度量多维尺度法是一种将多维空间的研究对象 (样本或变量)简化到低维空间进行定位、分析和归类, 同时又保留对象间原始关系的数据分析方法。 适用于无法获得研究对象间精确的相似性或相异性数据,仅能得到他们之间等级(秩次)关系数据的情形。 我们使用R语言vegan[18]包,基于Bray-Curtis 距离来进行 NMDS 分析(metaMDS函数), 同时使用R语言vegan[18]包, 基于 Bray-Curtis 距离来进行PERMANOVA分析(即adonis), 以检验分组间病毒群落结构的差异是否显著。

图3-4-20 NMDS 分析结果
- 一个点代表一个样本,点与点空间距离的排序关系近似物种组成结构的差异程度(Bray-Curtis 距离)的排序关系;
- 图中PERMANOVA分析的p值用于检验分组间病毒群落结构的差异是否显著
- 通常认为stress<0.2时有一定的解释意义;当stress<0.1时,可认为是一个好的排序;当 stress<0.05时,则具有很好的代表性。
3.5 功能分析
3.5.1 功能注释
05.Function/
├── 01.GenePredict
│ ├── combined_contig.faa [基因对应的蛋白序列]
│ ├── combined_contig.ffn [基因序列]
│ ├── combined_contig.gff [基因在contig上的位置信息]
│ └── quast [基因序列quast质量评估报告文件夹]
│ └── report.html [基因序列quast质量评估报告,文件夹中其他文件为报告的数据文件,可忽略]
├── 02.GeneAbundance
│ ├── gene_count.xls [基因fragments(read pairs)数表]
│ ├── gene_fpkm.xls [基因FPKM丰度表]
│ └── gene_tpm.xls [基因TPM丰度表]
├── KEGG [KEGG数据库功能分析文件夹]
│ ├── Abundance [KEGG数据库功能丰度分析文件夹]
│ │ ├── kegg_abundance_mixed.xls [KEGG数据库混合分层功能丰度表]
│ │ ├── kegg_abundance_stratified.xls [KEGG数据库根据物种来源分层的功能丰度表]
│ │ ├── kegg_abundance_unstratified.xls [KEGG数据库未分层的功能丰度表]
│ │ ├── kegg_ko_abundance_mixed.xls [KEGG ORTHOLOGY(KO)混合分层功能丰度表]
│ │ ├── kegg_ko_abundance_stratified.xls [KEGG ORTHOLOGY(KO)根据物种来源分层的功能丰度表]
│ │ ├── kegg_ko_abundance_unstratified.xls [KEGG ORTHOLOGY(KO)未分层的功能丰度表]
│ │ ├── kegg_pathway_abundance_mixed.xls [KEGG 通路混合分层功能丰度表]
│ │ ├── kegg_pathway_abundance_stratified.xls [KEGG 通路根据物种来源分层的功能丰度表]
│ │ └── kegg_pathway_abundance_unstratified.xls [KEGG 通路未分层的功能丰度表]
│ ├── GeneCount [gene计数分析文件夹]
│ │ ├── kegg_count_mixed.xls [KEGG数据库混合分层gene计数表]
│ │ ├── kegg_count_stratified.xls [KEGG数据库根据物种来源分层的gene计数表]
│ │ ├── kegg_count_unstratified.xls [KEGG数据库未分层的gene计数表]
│ │ ├── kegg_ko_count_mixed.xls [KEGG ORTHOLOGY(KO)混合分层gene计数表]
│ │ ├── kegg_ko_count_stratified.xls [KEGG ORTHOLOGY(KO)根据物种来源分层的gene计数表]
│ │ ├── kegg_ko_count_unstratified.xls [KEGG ORTHOLOGY(KO)未分层的gene计数表]
│ │ ├── kegg_pathway_count_mixed.xls [KEGG 通路混合分层gene计数表]
│ │ ├── kegg_pathway_count_stratified.xls [KEGG 通路根据物种来源分层的gene计数表]
│ │ └── kegg_pathway_count_unstratified.xls [KEGG 通路未分层的gene计数表]
│ └── kegg_function_annot.xls [gene KEGG数据库的比对和注释结果]
└── Uniprot [UniProt数据库功能分析文件夹]
├── Abundance [Uniprot数据库功能丰度分析文件夹]
│ ├── uniprot_abundance_mixed.xls [UniProt数据库混合分层功能丰度表]
│ ├── uniprot_abundance_stratified.xls [UniProt数据库根据物种来源分层的功能丰度表]
│ ├── uniprot_abundance_unstratified.xls [UniProt数据库未分层的功能丰度表]
│ ├── uniprot_keyword_abundance_mixed.xls [UniProt keywords混合分层功能丰度表]
│ ├── uniprot_keyword_abundance_stratified.xls [UniProt keywords根据物种来源分层的功能丰度表]
│ └── uniprot_keyword_abundance_unstratified.xls [UniProt keywords未分层的功能丰度表]
├── GeneCount [gene计数分析文件夹]
│ ├── uniprot_count_mixed.xls [UniProt数据库混合分层gene计数表]
│ ├── uniprot_count_stratified.xls [UniProt数据库根据物种来源分层的gene计数表]
│ ├── uniprot_count_unstratified.xls [UniProt数据库未分层的gene计数表]
│ ├── uniprot_keyword_count_mixed.xls [UniProt keywords混合分层gene计数表]
│ ├── uniprot_keyword_count_stratified.xls [UniProt keywords根据物种来源分层的gene计数表]
│ └── uniprot_keyword_count_unstratified.xls [UniProt keywords未分层的gene计数表]
└── uniprot_function_annot.xls [gene UniProt数据库的比对和注释结果]
基因预测
对于结合基于参考序列(Confirmed)和Denovo的方法获得的病毒contig序列, 我们使用 Prokka[12]软件进行基因预测, 获得每条contig上的gene序列(combined_contig.ffn)和蛋白序列(combined_contig.faa)。
UniProt 功能注释
使用 blastp[4]软件把 gene 的蛋白序列 (combined_contig.faa)与 UniProtKB/Swiss-Prot 数据库的病毒蛋白序 列( ViralZone, reviewed protein)比对, 筛选 e < 1e-3 的 best hit (e value 最小)比对结果,获取病毒基因蛋白的功能信息; 同时,根据gene与contig的对应关系(combined_contig.gff), 用对应contig的病毒分类作为gene的物种来源(uniprot_function_annot.xls)。
uniprot_function_annot.xls各列解释如下:
- qseqid: 查询序列ID(gene ID)
- sseqid: 比对到的数据库序列ID(UniProt数据库的蛋白ID)
- identity: identity=比对长度/查询序列长度与数据库序列长度的较小值,可理解为相似度
- length: 比对长度
- mismatch: 错配碱基数
- gapopen: 比对区域的gap数目
- qstart: 比对区域在查询序列(qseqid)上的起始位点
- qend: 比对区域在查询序列(qseqid)上的终止位点
- sstart: 比对区域在数据库序列(sseqid)上的起始位点
- send: 比对区域在数据库序列(sseqid)上的终止位点
- evalue: 随机序列中,存在比该数据库序列更好的匹配序列的概率,越低该比对结果越可靠
- bitscore: 比对得分,越高越可靠
- Contig: gene对应的contig ID
- Species: contig对应的病毒分类
KEGG 功能注释
KEGG(https://www.kegg.jp/)是了解高级功能和生物系统(如细胞、生物和生态系统), 从分子水平信息,尤其是大型分子数据集生成的基因组测序和其他高通量实验技术的实用程 序数据库。
使用 diamond[11] 软件 gene 的蛋白序列(combined_contig.faa)与 KEGG 的病毒蛋白数据库 (KEGG Virus)进行基因的同源性比对 (设定阈值为 e-value<=0.001)。由于每一条序列比对结果可能不止一条,为保证后续研究的 生物意义,从每条序列的 比对结果中筛选出 e-value 最低的作为该序列的比对结果。 同时,根据gene与contig的对应关系(combined_contig.gff), 用对应contig的病毒分类作为gene的物种来源(kegg_function_annot.xls)。
kegg_function_annot.xls各列解释如下:
- qseqid: 查询序列ID(gene ID)
- sseqid: 比对到的数据库序列ID(KEGG Virus数据库的gene ID)
- identity: identity=比对长度/查询序列长度与数据库序列长度的较小值,可理解为相似度
- length: 比对长度
- mismatch: 错配碱基数
- gapopen: 比对区域的gap数目
- qstart: 比对区域在查询序列(qseqid)上的起始位点
- qend: 比对区域在查询序列(qseqid)上的终止位点
- sstart: 比对区域在数据库序列(sseqid)上的起始位点
- send: 比对区域在数据库序列(sseqid)上的终止位点
- evalue: 随机序列中,存在比该数据库序列更好的匹配序列的概率,越低该比对结果越可靠
- bitscore: 比对得分,越高越可靠
- Contig: gene对应的contig ID
- Species: contig对应的病毒分类
基因计数
对于每个数据库(KEGG, UniProt)的比对结果,我们统计比对到每个数据库功能类别的gene序列数量, 即为该功能类别的基因计数(*_count_unstratified.xls)。 依据gene所属contig的病毒分类,计算根据物种来源分层的基因计数表(*_count_stratified.xls)。 所得计数表将用于后续可视化分析。
基因丰度
对于预测得到的gene序列(combined_contig.ffn), 我们通过如下步骤计算病毒gene的丰度, 并依据gene各个数据库(KEGG, UniProt)的功能注释结果, 计算每个数据库的每种功能类别在各样本的丰度(TPM), 用于后续可视化分析。
- 使用 BWA[2](默认参数: mem -k 30)软件,将去除污染后的 clean reads 分 别与gene序列(combined_contig.ffn)比对,过滤比对长度低于 reads 总长 80%的比对结果,统计gene的 fragments (read pairs)数(gene_count.xls);
- 计算每个gene 的 TPM 值(gene_tpm.xls), FPKM和TPM的计算请参照 文献中的公式
- 依据gene各个数据库(KEGG, UniProt)的功能注释结果, 计算每个数据库的每种功能类别在各样本的丰度,即将属于同一功能类别的基因 TPM(云流程可选择FPKM, fragments数)加和(*_abundance_unstratified.xls);
- 依据gene所属contig的病毒分类,计算根据物种来源分层的功能丰度表(*_abundance_stratified.xls)
所得丰度表将用于后续可视化分析。
3.5.2 基因计数可视化
05.Function/ [功能分析文件夹]
└── [KEGG或者Uniprot数据库]
├── GeneCount [gene计数分析文件夹]
│ ├── bar [gene计数柱形图文件夹]
│ │ └── [数据库功能分类方式]
│ │ └── Count_bar.svg [各功能类别gene计数柱形图]
│ ├── pie [gene计数占比饼图文件夹]
│ │ └── [数据库功能分类方式]
│ │ └── Count_pie.svg [各功能类别gene计数占比饼图]
│ ├── *_count_mixed.xls [参考3.5.1小节解读]
│ ├── *_count_stratified.xls [参考3.5.1小节解读]
│ └── *_count_unstratified.xls [参考3.5.1小节解读]
└── uniprot_function_annot.xls [参考3.5.1小节解读]
结果文件夹中有很多数据库功能分类方式的结果,这里只挑一部分说明,其它结果解读类似。
柱形图
柱形图用于展示每个功能类别的gene数量。
UniProtKB Keywords 是UniProt数据库的一种功能分类方式,其中包含了聚合酶(polymerase)的分类。

图3-5-1 各UniProtKB Keyword的gene数量柱形图
若功能类别超过20个,只展示gene数量最高的前20个。
饼图
饼图用于展示每个功能类别的gene数量占比。若类别数目超过20个,只展示gene数量占比最高的前20个

图3-5-2 各UniProtKB Keyword的gene数量占比饼图
3.5.3 功能丰度可视化
05.Function/ [功能分析文件夹]
└── [KEGG或者Uniprot数据库]
├── uniprot_function_annot.xls [参考3.5.1小节解读]
└── Abundance
├── *_abundance_mixed.xls [参考3.5.1小节解读]
├── *_abundance_stratified.xls [参考3.5.1小节解读]
├── *_abundance_unstratified.xls [参考3.5.1小节解读]
├── 01.BasicStatistics [基础统计文件夹]
│ ├── Barplots [百分比堆积柱形图文件夹]
│ │ └── [分组方案名]
│ │ └── [数据库功能分类方式]
│ │ ├── barplot.svg [百分比堆积柱形图]
│ │ └── table.xls [百分比堆积柱形图数据]
│ └── Heatmaps [丰度热图文件夹]
│ └── [分组方案名]
│ └── [数据库功能分类方式]
│ ├── heatmap.svg [丰度热图]
│ └── table.xls [丰度热图数据]
├── 02.GroupComparison [差异比较文件夹]
│ ├── ANOVA_Duncan [ANOVA和Duncan检验结果文件夹]
│ │ └── [分组方案名]
│ │ └── [数据库功能分类方式]
│ │ ├── [分组方案名]_all_feature_anova_results.xls [ANOVA检验结果]
│ │ ├── [分组方案名]_all_significant_feature_duncan_results.xls [Duncan检验结果, ANOVA无显著可缺失]
│ │ ├── [分组方案名]_all_significant_feature_barplot_of_duncan.svg [Duncan检验结果图, ANOVA无显著可缺失]
│ │ └── final_data_for_anova.xls [检验数据(原始丰度log转化,标准化之后)]
│ ├── DunnTest [Kruskal-Wallis和Dunn’s Test检验结果文件夹]
│ │ └── [分组方案名]
│ │ └── [数据库功能分类方式]
│ │ └── DunnTest.xls [Kruskal-Wallis和Dunn’s Test检验结果表格]
│ └── LEfSe [LEfSe分析结果文件夹]
│ └── [分组方案名]
│ └── [数据库功能分类方式]
│ ├── data_for_lefse.xls [LEfSe分析输入文件]
│ ├── lefse.LDA.xls [LEfSe分析结果表格]
│ ├── lefse.normalized.txt [LEfSe标准化后的数据,中间文件]
│ ├── lefse.svg [LEfSe分析结果LDA图]
│ └── SignificantFeatures [差异功能物种来源分层柱形图文件夹]
│ ├── Q5UQG2_stratification_bar.svg [差异功能物种来源分层柱形图]
│ └── table_for_stratification_bar.txt [差异功能物种来源分层柱形图输入表]
└── 03.Correlation [相关性分析结果文件夹]
├── CorrelationHeatmap [相关性热图结果文件夹]
│ └── [数据库功能分类方式]
│ ├── Correlation_heatmap.svg [相关性热图]
│ ├── p_value_matrix.xls [相关系数p值矩阵]
│ └── spearman_rank_correlation_matrix.xls [spearman相关系数矩阵]
└── RDA [CCA/RDA结果文件夹]
└── [分组方案名]
└── [数据库功能分类方式]
├── [分组方案名]_RDA.envfit.xls [RDA各环境因子坐标,解释方差,p值]
├── [分组方案名]_RDA.Factors.PERMANOVA.xls [所有环境因子对病毒群落变异的解释方差的p值计算结果]
├── [分组方案名]_RDA_features_location_plot.svg [RDA病毒点图]
├── [分组方案名]_RDA.features.xls [RDA病毒点坐标]
├── [分组方案名]_RDA.Group.PERMANOVA.xls [分组方式对病毒群落变异的解释方差的p值计算结果]
├── [分组方案名]_RDA_sample_location_plot.svg [RDA样本点图]
├── [分组方案名]_RDA_sample_location_plot_with_labels.svg [RDA样本点图(带样本名)]
├── [分组方案名]_RDA.sample.xls [RDA样本点坐标]
└── DCA_output.xls [DCA分析各排序轴值]
该部分结果与3.4.3小节(病毒丰度可视化)大体相同,请参照3.4.3小节解读功能丰度可视化结果。
这里需要特别说明的是,在LEfSe分析之后,我们挑出存在差异的功能(LDA>2), 用物种来源分层柱形图展示差异功能的物种来源,如图3-5-3

图3-5-3 KEGG数据库差异功能物种来源分层柱形图
3.6 噬菌体宿主预测
06.HostPredict/ └── combined_contig_predicted_host.xls [噬菌体宿主预测结果]
使用 CRISPR Recognition Tool( CRT, http://www.room220.com/crt/)从 Refseq 数据库的 细菌基因组中构建 CRISPR-Cas spacer 数据库, 然后使用 blastn-short[4]与 前述结合Denovo方法和基于参考序列方法鉴定的病毒序列(combined_contig.fa)比对, 获取噬菌体可能的宿主信息。由于目前已知的 spacer 序列有限,因此只有 一部分病毒 contigs 能预测出可能的宿主。
筛选标准如下:
- e-value<=1e-10,比对相似性(identity)>=95%;
- 选取 best hit (e-value最小) 比对结果;
- 统计宿主种水平信息。
combined_contig_predicted_host.xls表格各列解释如下:
- #Contig ID: Contig标识;
- Predicted Host: 预测得到的宿主,多个宿主用“+”隔开。
四 分析所用软件的版本
| 软件 | 版本 |
|---|---|
| Trimmomatic | 0.39 |
| BWA | 0.7.17 |
| Megahit | 1.2.9 |
| blast | 2.12.0+ |
| CDHIT | 4.8.1 |
| QUAST | 5.0.2 |
| MetaGeneMark | 3.38 |
| hmmsearch | 3.3.2 |
| diamond | 0.9.14 |
| Prokka | 1.14.6 |
| LEfSe | 1.0.8 |
| dunn.test | 1.3.5 |
| ggpubr | 0.2.4 |
| agricolae | 1.3.3 |
| pheatmap | 1.0.12 |
| vegan | 2.6.0 |
| ggplot2 | 3.3.5.9000 |
| qiime2 | qiime2-2022.2 |
| ape | 5.3 |