微科盟宏基因组数据分析结题报告
一 概述
微生物世界是分子多样性最大的天然资源库,基于菌株水平的 传统分离培养技术为人们认识微生物多样性提供了可能, 但是据估计自然界中超过99%的微生物不能 通过传统的分离培养技术获得其纯培养,从而导致环境微生物中的多样性 基因资源难以被发现。许多重要的微生物我们还不能识别, 随着微生物活性产物的广泛研究和深入开发利用, 从环境微生物中筛选到新活性物质的几率将逐步下降。 而如何开拓利用环境微生物新资源是微生物研究的重要课题。 为此研究者们开发了多种以特定环境微生物为研究对象的高通量测序方法。
宏基因组学(Metagenomics),是一种直接对微生物群体中包含的全部基因组信息 进行研究的手段。宏基因组学绕过对微生物个体进行分离培养, 应用基因组学技术对自然环境中的微生物群落进行研究的一门学科。 它规避了对样品中的微生物进行分离培养,提供了一种对不可分离培养的微生物 进行研究的途径,更真实的反应样本中微生物组成、互作情况,同时 在分子水平对其代谢通路、基因功能进行研究。近年来,随着测序技术和信息技术 的快速发展,利用新一代测序技术(Next Generation Sequencing)研究 Metagenomics, 能快速准确的得到大量微生物基因数据和丰富的微生物研究信息, 从而成为研究微生物多样性和群落特征的重要手段。细菌基因组相对较小, 通常仅有一条环状DNA和质粒,通过高通量测序,可以了解其全部遗传信息。 这也已经成为微生物研究的重要手段之一,为细菌的遗传进化、疾病预防与治疗、 疫苗与抗生素的开发等提供重要的信息。如致力于研究微生物与人类疾病健康关系的 人体微生物组计划(HMP, Human Microbiome Project, http://www.hmpdacc.org/ ), 研究全球微生物组成和分布的全球微生物组计划(EMP, Earth Microbiome Project, http://www.earthmicrobiome.org/ )都主要利用高通量测序技术进行研究。
二 项目流程
2.1 实验流程

图2-1 实验工作流程图
2.2 数据分析流程
分析流程简要步骤如图2-2所示,详细步骤见 结果解读部分

图2-2 信息分析流程图
三 结果文件夹解读
3.1 序列质控和去宿主(../00-QCStats)
|--00-QCStats
| |--1-QC_report_Rawfastq/*.html [原始数据fastqc质检结果]
| |--2-QC_report_Filtered/*.html [序列质控和去宿主序列之后的fastqc质检结果]
| |--reads_summary.txt [数据产出质量情况一览表]
本项目采用Illumina Novaseq平台对测序样本进行双端测序。 基于FASTQ格式的测序文件是一种存储序列信息的特定文件,推荐用Notepad++等文本编辑器或者在电脑终端中打开。 FASTQ文件每四行对应一条测序Read:第一行以符号“@”起始,对应于序列ID和相应的描述信息; 第二行为实际测得的碱基序列;第三行以符号“+”起始; 而第四行的字符串则记录了第二行序列中每个碱基所对应的测序质量。
采用 Illumina测序平台测序获得的原始数据(Raw Data)存在一定比例低质量数据,
为了保证后续分析的结果准确可靠,需要对原始的测序数据进行预处理,包括质控
(Trimmomatic[6]
参数:ILLUMINACLIP:adapters_path:2:30:10 SLIDINGWINDOW:4:20 MINLEN:50),
和去宿主序列(Bowtie2[7]参数:--very-sensitive),获取用于后续分析的有效序列(clean data)。
测序数据预处理统计结果见表 3-1。序列质控步骤关键参数解释如下:
- 去除接头序列 (参数ILLUMINACLIP:adapters_path:2:30:10);
- 扫描序列(4bp滑窗大小),如果平均质量分低于20(99%正确率),切除后续序列(参数SLIDINGWINDOW:4:20);
- 去除最终长度小于50bp的序列(参数MINLEN:50)。
表3-1 数据产出质量情况一览表
| Sample ID | InsertSize(bp) | SeqStrategy | RawReads(#) | Raw Base(GB) | %GC | Raw Q20(%) | Raw Q30(%) | Clean Reads(#) | Cleaned(%) | Clean Q20(%) | Clean Q30(%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 | 350 | (150:150) | 23613034 | 7.08 | 61 | 97.70 | 93.78 | 22416119 | 94.93 | 98.80 | 95.62 |
| C2 | 350 | (150:150) | 27584954 | 8.28 | 61 | 97.10 | 92.62 | 26060963 | 94.48 | 98.55 | 94.94 |
| C3 | 350 | (150:150) | 23777022 | 7.13 | 61 | 98.45 | 95.71 | 22889304 | 96.27 | 99.27 | 97.08 |
| C4 | 350 | (150:150) | 27007788 | 8.10 | 61 | 97.63 | 93.65 | 25636029 | 94.92 | 98.77 | 95.52 |
| T11 | 350 | (150:150) | 29809022 | 8.94 | 62 | 97.62 | 93.48 | 28346006 | 95.09 | 98.63 | 95.16 |
| T12 | 350 | (150:150) | 24702558 | 7.41 | 61 | 97.72 | 93.78 | 23570823 | 95.42 | 98.72 | 95.43 |
| T13 | 350 | (150:150) | 28274974 | 8.48 | 61 | 97.63 | 93.54 | 26703496 | 94.44 | 98.69 | 95.32 |
| T21 | 350 | (150:150) | 25016746 | 7.51 | 60 | 97.74 | 93.74 | 23831131 | 95.26 | 98.70 | 95.35 |
| T22 | 350 | (150:150) | 24980036 | 7.49 | 60 | 97.55 | 93.21 | 23555813 | 94.30 | 98.57 | 94.96 |
| T23 | 350 | (150:150) | 24026221 | 7.21 | 62 | 97.64 | 93.58 | 22758509 | 94.72 | 98.69 | 95.34 |
- Sample ID: 样品名;
- InsertSize(bp): InsertSize是在建库切胶时选择的长度,合适的InsertSize能避免测序接头污染;
- SeqStrategy: 测序策略(一般为双端,各150bp);
- RawReads(#):测序Raw reads的数量;
- Raw Base(GB): 以GB为单位的Raw reads数量, 测序原始数据的总碱基数,即为Raw reads数量乘以测序长度算得;
- %GC:G/C碱基数占总碱基数量的百分比;
- Clean Reads(#):过滤(质控和去宿主序列)后获得的Clean reads 数量;
- Cleaned(%):过滤后剩余的序列数占Raw reads 的百分比;
- Q20: 质量分高于20的碱基所占比例;
- Q30,质量分高于30的碱基所占比例。
3.2 物种注释(1-TaxaAundanceAnalysis)
1-TaxaAundanceAnalysis ├── 1-AbundanceSummary │ ├── 1-RelativeAbundance │ │ └── otu_table.[分类水平].relative.txt [对应分类水平相对丰度表] │ ├── classified_stat_relative.csv [样本各水平物种注释比例统计表] │ └── Classified_stat_relative.svg [样本各水平物种注释比例统计图] └── All.Taxa.OTU.txt [物种reads数丰度总表, 第一列为NCBI taxid, 不是OTU, 列名OTU只是格式需要]
为了研究样品物种组成及多样性信息,使用Kraken2 [4] 将各样本质控和去宿主之后的序列和自建的微生物核酸数据库 (筛选NCBI NT核酸数据库和RefSeq全基因组数据库中属于细菌, 真菌,古菌,病毒的序列)比较(参数为--confidence 0.2) , 计算样本中所含有物种的序列数。 Kraken2是基于Kraken1后研发的的最新版本(2018)。 Kraken系列软件是一种使用精确k-mer匹配的分类系统,可实现高精度和快速分类速度。 该分类器将查询序列中的每个k聚体与包含给定k聚体的所有基因组的最低共同祖先(LCA)匹配。 Kraken2在Kraken1的基础上提供了重大改进, 达到了数据库构建时间更短,分类速度更快的效果。 在使用Kraken对宏基因组测序数据进行物种分类之后, 我们继续用Bracken[5](默认参数) 对Kraken2得到的分类结果进行分类后贝叶斯重新估算丰度来估算 宏基因组样本的种水平丰度。
基于Bracken结果的绝对丰度(reads 数)及注释信息, 对每个样品在一共7个分类水平界门纲目科属种 (Kingdom,Phylum,Class,Order,Family,Genus,Species) 上的序列数目占总序列数的比例进行统计, 可以有效的评估样本的物种注释分辨率 (注释到属/种的比例越高表示样本的注释效果越好), 下图展示了每个样本中在各分类水平注释的相对程度:

图3-2-1 各个样品在各分类水平上的序列注释程度柱形图
说明:横坐标(Sample Name)是样品名,纵坐标(Sequence Number Percent)表示注释到该水平的序列数目占总注释数据的比率, 柱状图自上而下的颜色顺序对应于右侧的图例颜色顺序。 每个分类水平最高值为1,代表100%的序列都得到了至少在这个级别的注释。
经过分析和统计,您样本中,检测到的物种有:Archaea(64146), Bacteria(18490333), Bamfordvirae(12), Fungi(6642), Heunggongvirae(2087), Loebvirae(1), Pararnavirae(96), Sangervirae(4), Shotokuvirae(28), Viruses(35); 物种占比情况:Archaea(0.35%), Bacteria(99.61%), Bamfordvirae(0.00%), Fungi(0.04%), Heunggongvirae(0.01%), Loebvirae(0.00%), Pararnavirae(0.00%), Sangervirae(0.00%), Shotokuvirae(0.00%), Viruses(0.00%)。
宏基因组物种注释得到的丰度,建议只在样本/分组之间比较, 不要在物种之间比较。扩增子测序得到的优势物种, 在宏基因组中,丰度不一定很高,甚至可能检测不到, 主要有以下四点原因:
- 扩增子是用引物定向扩增特征序列的, 定向意味着不会受其它的DNA序列干扰, 比如扩增真菌的18S序列,不会受到细菌的16S和宿主序列干扰, 扩增16S序列,不会受到宿主基因干扰,也不会受到真菌干扰; 相比之下,宏基因组不对提取的DNA进行筛选, 只是打碎后,测所有的序列,这意味着,虽然您可能只关注真菌,但由于没有对DNA进行筛选, 您也会测到大量的细菌序列,宿主序列等干扰序列, 总的测序量是不变的,如果样本中细菌比较多,测到的真菌就会相对比较少;
- 扩增特征序列意味着扩增得到的序列大部分都是能够准确注释到物种名称的; 相比之下,宏基因组的优势在于注释功能,宏基因组测序得到的大部分序列都不是特征序列, 也就是说这些序列在很多物种里都有,没办法确定它来自哪个物种;
- 宏基因组的物种定量还受到基因组大小影响, 一个用扩增子手段得到的优势物种,如果它的基因组相对较小, 那么它在宏基因组测序中被测到的几率就小很多,因此丰度不一定高,甚至可能没有;
- 宏基因组的物种定量也受到数据库的影响,目前,还有很多物种的基因组是未知的, 一个知道全基因组的物种相比一个只知道部分核酸序列的物种, 在宏基因组定量分析中,是存在明显优势的, 全基因组比对到的reads肯定比几条核酸序列比对到的reads多, 微科盟的物种定量数据库,采用RefSeq全基因组数据库 + NCBI NT核酸数据库,已经足够全面了。
用宏基因组进行物种定量分析,与扩增子相比,是存在明显劣势的, 建议主要用扩增子进行多样性分析,而用宏基因组进行功能分析, 宏基因组的物种定量结果,以及扩增子的功能预测结果都不够准确, 只能作为参考
3.3 物种可视化(1-TaxaAundanceAnalysis)
1-TaxaAundanceAnalysis ├── 1-AbundanceSummary │ ├── 1-RelativeAbundance │ │ └── otu_table.[物种分类水平].relative.txt [对应分类水平相对丰度表] │ ├── 2-Barplots │ │ ├── All.Taxa.OTU.taxa-bar-plots │ │ │ ├── * [文件夹内其它文件为报告数据文件,无需关注] │ │ │ └── Summary_请点此文件查看.html [总reads数>=10的物种(taxid水平)qiime2 百分比堆积柱形图报告] │ │ ├── All.Taxa.OTU.taxa-bar-plots.1000 │ │ │ ├── * [文件夹内其它文件为报告数据文件,无需关注] │ │ │ └── Summary_请点此文件查看.html [总reads数>=1000的物种(taxid水平)qiime2 百分比堆积柱形图报告] │ │ ├── Barplot-of-Group-Mean │ │ │ ├── [分组方案名]_[物种分类水平]_mean_barplot.svg [对应水平总丰度前20物种分组平均丰度百分比堆积柱形图] │ │ │ └── [分组方案名]_[物种分类水平]_mean_table.xls [对应水平总丰度前20物种分组平均丰度百分比统计表] │ │ └── Taxa-bar-plots-top20 │ │ ├── [物种分类水平]_[分组方案名]_barplot.svg [对应水平总丰度前20物种丰度百分比堆积柱形图] │ │ └── [物种分类水平]_[分组方案名]_table.xls [对应水平总丰度前20物种丰度百分比统计表] │ ├── 3-Heatmaps │ │ ├── Heatmap_top20 │ │ │ └── [物种分类水平] │ │ │ ├── [分组方案名]_group_mean_heatmap.svg [对应水平总丰度前20物种分组平均(log10(绝对丰度+1))热图] │ │ │ ├── [分组方案名]_group_mean_table.xls [对应水平总丰度前20物种分组平均(log10(绝对丰度+1))统计表] │ │ │ ├── [分组方案名]_heatmap.svg [对应水平总丰度前20物种log10(绝对丰度+1)热图] │ │ │ └── [分组方案名]_table.xls [对应水平总丰度前20物种log10(绝对丰度+1)统计表] │ │ └── Heatmap_top20_clustered │ │ └── [物种分类水平] │ │ ├── [分组方案名]_heatmap.svg [对应水平总丰度前20物种log10(绝对丰度+1)聚类热图] │ │ └── [分组方案名]_table.xls [对应水平总丰度前20物种log10(绝对丰度+1)统计表] │ ├── classified_stat_relative.csv [样本各水平物种注释比例统计表] │ └── Classified_stat_relative.svg [样本各水平物种注释比例统计图] ├── 2-AbundanceComparison │ ├── ANCOM │ │ ├── [分组方案名]-ANCOM-[物种分类水平] │ │ │ └── Summary_请点此文件查看.html [ANCOM分析报告] │ │ └── [分组方案名]-ANCOM-[物种分类水平].qzv [ANCOM分析报告qzv格式, 可拖拽至https://view.qiime2.org/查看] │ ├── DunnTest │ │ ├── [物种分类水平]_DunnTest.xls [Dunn's Test多重比较结果表] │ │ └── [物种分类水平]_relative_abundance.xls [Dunn's Test输入表] │ ├── LEfSe │ │ └── [物种分类水平] │ │ ├── [分组方案名]_[物种分类水平]_lefse_LDA2.cladogram.svg [LDA>2的LEfSe结果cladogram图] │ │ ├── [分组方案名]_[物种分类水平]_lefse_LDA2.LDA.xls [LDA>2的LEfSe结果表] │ │ ├── [分组方案名]_[物种分类水平]_lefse_LDA2.lefseinput.xls [LEfSe对输入丰度表进行标准化后的丰度] │ │ ├── [分组方案名]_[物种分类水平]_lefse_LDA2.svg [LDA>2的LEfSe结果柱形图] │ │ ├── [分组方案名]_[物种分类水平]_lefse_LDA4.cladogram.svg [LDA>4的LEfSe结果cladogram图] │ │ ├── [分组方案名]_[物种分类水平]_lefse_LDA4.LDA.xls [LDA>4的LEfSe结果表] │ │ ├── [分组方案名]_[物种分类水平]_lefse_LDA4.svg [LDA>4的LEfSe结果柱形图] │ │ └── [分组方案名]_[物种分类水平]_lefse.xls [LEfSe分析输入表] │ └── VennAndFlower │ ├── [分组方案名]_binary.xls [0-1表, 1代表有分组有该物种, 0代表无] │ ├── [分组方案名]_common_species.xls [各分组共有物种列表] │ ├── [分组方案名]_[分组]_special_species.xls [分组特有物种列表] │ └── [分组方案名]_Venn_plot.svg [Venn图] ├── 3-DiversityAnalysis [与云流程分步骤有细微差异,因为抽平结果不一样] │ ├── bray_curtis_distance_matrix.xls [Bray-Curtis距离矩阵] │ ├── bray_curtis_emperor │ │ ├── * [文件夹内其它文件为报告数据文件,无需关注] │ │ └── Summary_请点此文件查看.html [qiime2基于Bray-Curtis距离的PCoA分析结果报告] │ ├── nmds_points_ordinates.xls [NMDS散点图中各点坐标] │ ├── NMDS.svg [NMDS散点图] │ ├── PCoA_3D.svg [PCoA 3D散点图] │ ├── pcoa_points_ordinates.xls [PCoA 散点图中各点坐标] │ └── PCoA.svg [PCoA 2D散点图] ├── 4-CorrelationAnalysis │ ├──CorrelationHeatmap │ │ └── [物种分类水平] │ │ ├── Correlation_heatmap.svg [相关性热图] │ │ ├── p_value_matrix.xls [相关系数p值矩阵] │ │ └── spearman_rank_correlation_matrix.xls [spearman相关系数矩阵] │ └── RDA │ └── [物种分类水平] │ ├── [分组方案名]_RDA.envfit.xls [RDA各环境因子坐标,解释方差,p值] │ ├── [分组方案名]_RDA.Factors.PERMANOVA.xls [所有环境因子对微生物群落变异的解释方差的p值计算结果] │ ├── [分组方案名]_RDA_features_location_plot.svg [RDA微生物点图] │ ├── [分组方案名]_RDA.features.xls [RDA微生物点坐标] │ ├── [分组方案名]_RDA.Group.PERMANOVA.xls [分组方式对微生物群落变异的解释方差的p值计算结果] │ ├── [分组方案名]_RDA_sample_location_plot.svg [RDA样本点图] │ ├── [分组方案名]_RDA_sample_location_plot_with_labels.svg [RDA样本点图(带样本名)] │ ├── [分组方案名]_RDA.sample.xls [RDA样本点坐标] │ └── DCA_output.xls [DCA分析各排序轴值] ├── All.Taxa.OTU.txt [物种reads数丰度总表, 第一列为NCBI taxid, 不是OTU, 列名OTU只是格式需要] └── keep_[物种名列表] [只保留对应物种的分析结果文件夹,文件夹解读同上1-TaxaAundanceAnalysis]
3.3.1 物种统计( 1-TaxaAundanceAnalysis/1-AbundanceSummary )
图3-3-1展示了样本中物种在不同分类水平相对分布情况的柱形图:
图3-3-1 各个样本在各分类水平的物种相对分布情况的柱形图(全部物种,点击 此处 打开新窗口查看 )
说明:横坐标(Sample Name)是样品名, 纵坐标(Relative Abundance)表示相对丰度。 其他分类水平的物种相对丰度图也都可以通过交互式网页打开, 并对样本或者物种在图片中呈现的顺序根据元数据(包括分组信息) 或者相对丰度的大小进行调节。 Level 1,2,3,4,5,6,7依次代表分类水平界门纲目科属种。

图3-3-2 各个样品在门水平上的相对分布情况柱形图(相对丰度前20的物种)
说明:通常在种/属水平因为分类种类过多而在图例中 无法全部展示所有分类,特增加本图是对图3-3-1进行补充。 横坐标(Sample Name)是样品名, 纵坐标(Sequence Number Percent)表示注释 到该门水平的序列数目占总注释数据的比率, 柱状图自上而下的颜色顺序对应于右侧的图例颜色顺序。 在门水平没有注释的序列被归为unclassified一类。 图例中最多显示最优势的20个种类, 余下的相对丰度较低的物种被归类为Other在图中展示。 更多完整的物种分布情况请参见结果部分 1-TaxaAundanceAnalysis/1-AbundanceSummary/2-Barplots/ 。
除了通过网页版报告,用户也可以通过本地文件夹查看结果。 结果文件夹中包含的.qzv文件是Qiime2的专属可视化文件格式, 可以通过网页https://view.qiime2.org/ 进行交互性查看, 也可以直接点开相应的qzv解压缩文件夹通过网页形式直接点开HTML文件 “Summary_请点此文件查看.html”查看。 网页报告只选取了部分代表性信息用于引导用户阅读, 然而本地文件夹的结果目录中包含了大量其他有用的信息, 请用户自行查看。
为了研究不同样品间的相似性,还可以通过 对样品进行聚类分析从而构建样品的聚类。 选取关注的物种(默认选取物种绝对丰度排名前20), 根据样品的物种组成,实现样品聚类, 以此考察不同样品或者分组间的相似或差异性(此处为门水平分类的热图); 也根据物种丰度在各样本的分布进行聚类,寻找物种或样本的聚集规律。

图3-3-3 门分类水平的热图聚类结果
说明:纵轴为样品名称信息,同时也包括了分组信息。 横轴为物种注释名称(本图为门水平)。 图中上方的聚类树为物种在各样本中分布的相似度聚类, 左侧的聚类树为样品聚类树,中间的热图是物种的相对丰度热图, 颜色与相对丰度的关系见图上方的刻度尺。 其他分类等级完整的热图聚类结果请参见结果部分 1-TaxaAundanceAnalysis/1-AbundanceSummary/3-Heatmaps/ 。
3.3.2 物种差异 ( 1-TaxaAundanceAnalysis/2-AbundanceComparison )
LEfSe寻找每一个分组的特征微生物(默认为LDA>2的微生物) [9], 也就是相对于其他分组,在这个组中丰度较高的微生物 (如图3-3-4。完整的差异性分析结果请见结果部分 1-TaxaAundanceAnalysis/2-AbundanceComparison/LEfSe/ 。

图3-3-4 LEfSe分析LDA柱形图
说明:每一横向柱形体代表一个物种, 柱形体的长度对应LDA值,LDA值越高则差异越大。 柱形的颜色对应该物种是那个分组的特征微生物, 特征微生物(在对应分组中的丰度相对较高)。 当只有两个分组时,LDA在图中可能有正负之分, 正负只是为了区分分组,没有实际意义, 绝对值才代表差异程度

图3-3-5 属水平微生物LEfSe分析cladogram图
cladogram图由内到外,分别对应界门纲目科属种不同的分类层级,层级间连线代表所属关系。 每个圆圈节点代表一个微生物,节点为黄色代表在分组间差异不显著, 不为黄色则代表该微生物是对应颜色分组的特征微生物(在该分组丰度显著较高)。 有颜色的扇形区域标注了特征微生物的下属分类区间。
ANCOM(Analysis of composition of microbiomes) [10] 是专门用于比较微生物组学数据中物种在组间的显著性差异的分析方法。 ANCOM分析不依赖于数据的分布假设, 并解决了在其他方法中相对丰度分析所带来的限制, 从而能够有效降低结果的假阳性。 从下面链接中我们可以得知属水平分类的物种在组间表现了丰度的显著差异:
图3-3-6 属水平分类的ANCOM丰度比较结果
说明:在ANCOM分析中,W值是一个衡量组间差异显著性的统计量(类似F值,t值), W值越高,代表该物种在组间的差异显著性越高。 图中的每一个点都代表了一个比较的物种, 纵坐标代表W值,横坐标clr值代表组间样品丰度的差异程度, 数字越高代表相对丰度差异越大。因此在图中的点越靠近右上角, 则代表该物种与其他物种(大部分集中在左下角)相比更具有显著性差异。 组间具有差异显著的物种被列在下面相应的表格中。
在样本中,根据物种是否存在来寻找分组之间的特有或共有的物种, 对于分组较少(小于等于5)的实验方案,我们绘制韦恩图(Venn diagram) 分析不同样品组之间特有或共有的物种(种水平), 用于统计多个样本中所共有和独有的物种数目, 可以比较直观地表现样本分组间物种组成相似性及重叠情况(图3-3-7)。 对于分组较多的(大于等于3),我们绘制了花瓣图, 花瓣图中,花瓣里是对应分组特有的物种数目,中心是所有分组共有的物种数目。 同时也给出了特有和共有的taxa,丰度较高且特有的物种具有重要分析价值, 请参见 1-TaxaAundanceAnalysis/2-AbundanceComparison/VennAndFlower

3.3.3 物种多样性( 1-TaxaAundanceAnalysis/3-DiversityAnalysis )
目前适用于生态学研究的降维分析主要是主成分分析 (PCA,Principal Component Analysis) 和无度量多维标定法(NMDS,Non-Metric Multi-Dimensional Scaling)分析。 其中,PCA是基于线型模型的一种降维分析,它应用方差分解的方法对多维数据进行降维, 从而提取出数据中最主要的元素和结构;PCA 能够提取出最大程度反映样品间差异的两个坐标轴, 从而将多维数据的差异反映在二维坐标图上,进而揭示复杂数据背景下的简单规律。 而NMDS是非线性模型,其目的是为了克服线性模型的缺点,更好地反映生态学数据的非线性结构, 应用NMDS分析,根据样本中包含的物种信息,以点的形式反映在多维空间上, 而不同样本间的差异程度则是通过点与点间的距离体现,能够反映样本的组间或组内差异等。 基于不同分类层级的物种丰度表,我们进行了PCA和NMDS分析,如果样品的物种组成越相似, 则它们在PCA和NMDS图中的距离则越接近。 详细的数值可以查看结果目录 。
PCA是主坐标分析(PCoA)的一种特殊情况(基于欧式距离的PCoA就是PCA)。 本报告利用PCoA分析方法,从多维数据中 提取出最主要元素和能够最大程度反映样品间差异的三个坐标轴, 从而将多维数据的差异反映在三维坐标图上,进而揭示复杂数据背景下的简单规律。 我们基于Bray Curtis距离来进行PCoA分析,并选取贡献率最大的主坐标组合进行作图展示, 图中样品的距离越接近,表示样品的物种组成结构越相似(图3-3-8)。 分析结果文件夹分别提供了PCoA的2D和3D图,方便选择使用。
图3-3-8 基于Bray Curtis距离矩阵的的PCoA 3D图 (点击此处打开新窗口查看,图片可拖动旋转,可调色)
说明:横坐标(Axis 1)表示第一主成分, 百分比则表示第一主成分对样品差异的贡献值;纵坐标(Axis 2) 表示第二主成分,百分比表示第二主成分对样品差异的贡献值; Axis 3坐标表示第三主成分,百分比表示第三主成分对样品差异的贡献值。 客户可以根据元数据的分组信息可以个性化展示其他分类的距离关系。 举例:可在右边设置区域的Select a Color Category下拉单选择分组信息, 则同一个组的样品使用同一种颜色表示。
3.3.4 物种相关性( 1-TaxaAundanceAnalysis/4-CorrelationAnalysis )
相关性热图
如果提供了环境因子的数据,比如 pH值、温度值、临床因子等,可以进行相关性热图分析
相关性热图的绘制主要运用了R语言pheatmap包[11]。 相关性热图可以用于分析环境因子或其他临床表型数据与微生物群落或物种之间是否显著相关, 然后计算环境因子与微生物物种间的Spearman相关系数,并用热图展示。 该分析可以挑选出与某种环境因子或疾病显著相关的物种。

图3-3-9 微生物与表型之间的相关性热图
X轴上为环境因子,Y轴为物种。通过计算获得R值(秩相关)和p值。 R值在图中以不同颜色展示,p值若小于0.05则用 * 号标出, 右侧图例是不同R值的颜色区间,同时,左边的色条标注了物种所属门分类。 * 0.01≤ p <0.05,** 0.001≤p < 0.01,*** p < 0.001。 先依据显著的环境因子个数排名(越多越靠前),再依据显著的p值平均值排名(越低越靠前), 该图只展示排名前20个微生物,如果没有显著相关的微生物,则无法作图。
RDA/CCA分析
CCA/RDA的分析主要依赖R语言vegan包[12], 以及用ggplot2[13]进行可视化。 CCA/RDA(DCA判断用哪一种分析)分析是基于对应分析发展的一种排序方法, 将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析。 RDA是基于线性模型,CCA是基于单峰模型。本报告先进行DCA分析,看最大轴的值是否大于4,如果大于4.0, 就选CCA,否则选RDA。该分析主要用来反映微生物群落与环境因子之间的关系, 可以检测环境因子、样品、微生物群落(或功能)三者之间的关系或者两两之间的关系, 可得到影响样品分布的重要环境驱动因子。该分析给出的所有p值都是反映解释变量 (连续的环境因子,或者分组方案)对微生物群落变异的解释程度是否显著 (简单的说就是解释变量对微生物群落是否有影响,影响是否显著),所有p值都是用R语言vegan包里的置换检验得出的 (permutation test),*_features_location_plot图中的p值 反映了所有连续的数值变量(环境因子)对微生物差异的解释程度(总的p值), 表格*_RDA.envfit中的p值反映了每个环境因子对微生物差异的解释程度, *RDA_sample_location_plot图中的p值反映了分组对微生物差异的解释程度,p<0.05, 解释方差显著。

图3-3-10 种水平微生物CCA/RDA排序图
在CCA/RDA物种排序图内,环境因子用箭头表示, 箭头连线的长度代表某个环境因子与群落分布和种类分布间相关程度的大小(解释方差的大小), 箭头越长,说明相关性越大,反之越小。箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小, 夹角越小,相关性越高;反之越低。环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系, 钝角时呈负相关关系。每个点代表一个物种,点越大,对应物种丰度越高, 灰色点代表丰度较低的物种,未在图中注释物种名称,将物种投影到各个环境因子, 对应的值即为该物种倾向于存在的环境(喜欢的环境)。或者说,将物种点与原点连线, 物种间,物种与环境因子间,环境因子间的夹角的余弦值近似于相关系数,至于有多近似, 就要看RDA1/CCA1和RDA2/CCA2两个坐标轴的解释方差百分比有多大,越大越近似。 对于样本排序图,样本点之间的距离近似于微生物群落结构差异程度, 样本点投影到环境因子对应的值近似于该样本真实的环境因子值。

图3-3-11 CCA/RDA样本排序图
环境因子用箭头表示, 箭头连线的长度代表某个环境因子与群落分布和种类分布间相关程度的大小(解释方差的大小), 箭头越长,说明相关性越大,反之越小。箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小, 夹角越小,相关性越高;反之越低。环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系, 钝角时呈负相关关系。样本点之间的距离近似于微生物群落结构差异程度, 样本点投影到环境因子对应的值近似于该样本真实的环境因子值。
*_RDA.envfit.xls表格各列解释如下:
- 环境因子
- RDA1: 表征环境因子的方向,与横轴正方向夹角的余弦值
- RDA2: 表征环境因子的方向,与横轴正方向夹角的正弦值
- r2: 环境因子对微生物群落变异的解释方差
- p value: 置换检验计算的解释方差的p值,小于0.05,可推断该环境因子对微生物群落影响显著
*_RDA.*.PERMANOVA.xls表格为所有环境因子(或分组方式)对微生物群落结构变异的解释方差的p值计算结果, 用的是PERMANOVA置换检验的方法,各列解释如下:
- 方差组分(模型部分,剩余部分)
- Df: 自由度
- Variance: 方差剖分
- F: PERMANOVA检验统计量
- Pr(>F): PERMANOVA检验p值
3.4 功能注释( ../01-BaseTables , 2-FuctionAnalysis, 3-AMRAnalysis )
01-BaseTables/ ├── All.Kraken2.taxa.txt [Kraken2分析得到的物种丰度总表, 用于后续物种可视化] ├── All.Metaphlan2.taxa.txt [Metaphlan2分析得到的物种丰度总表, HUMAnN副产物, 物种较少, 不用于物种可视化] ├── All.UniRef90.genefamilies_stratified.tsv [UniProt数据库 UniRef90蛋白 物种来源分层 丰度表] ├── All.UniRef90.genefamilies.tsv [UniProt数据库 UniRef90蛋白 混合分层 丰度表, 相当于stratified+unstratified] └── All.UniRef90.genefamilies_unstratified.tsv[UniProt数据库 UniRef90蛋白 丰度表] 2-FuctionAnalysis/ ├── 1-KEGG │ ├── All.KEGG.Module.txt [KEGG数据库模块丰度表] │ ├── All.KEGG.Pathway.Level1.txt [KEGG数据库Level1层级通路丰度表] │ ├── All.KEGG.Pathway.Level2.txt [KEGG数据库Level2层级通路丰度表] │ ├── All.KEGG.Pathway.Level3.txt [KEGG数据库Level3层级通路丰度表] │ ├── All.KEGG.Pathway.txt [KEGG数据库通路(ID)丰度表] │ ├── All.KO.abundance_stratified.tsv [KEGG数据库 KO 物种来源分层 丰度表] │ ├── All.KO.abundance.tsv [KEGG数据库 KO 混合分层 丰度表, 相当于stratified+unstratified] │ ├── All.KO.abundance_unstratified.tsv [KEGG数据库 KO 丰度表] │ └── All.KO.detail.txt [KEGG数据库 KO 详细注释(最后几列) 丰度表] ├── 2-Metacyc │ ├── All.Metacyc.pathabundance_stratified.tsv [Metacyc数据库 通路 物种来源分层 丰度表] │ └── All.Metacyc.pathabundance_unstratified.tsv [Metacyc数据库 通路 丰度表] ├── 3-EggNOG │ ├── All.EGGNOG.abundance_stratified.tsv [EggNOG数据库 COG 物种来源分层 丰度表] │ ├── All.EGGNOG.abundance.tsv [EggNOG数据库 COG 混合分层 丰度表, 相当于stratified+unstratified] │ ├── All.EGGNOG.abundance_unstratified.tsv [EggNOG数据库 COG 丰度表] │ ├── All.EGGNOG.detail.txt [EggNOG数据库 COG 详细注释(最后几列) 丰度表] │ └── All.EGGNOG.Function.Category.txt [EggNOG数据库 COG 功能分类 丰度表] ├── 4-GO │ ├── All.GO.abundance_stratified.tsv [GO数据库 GO 物种来源分层 丰度表] │ ├── All.GO.abundance.tsv [GO数据库 GO 混合分层 丰度表, 相当于stratified+unstratified] │ ├── All.GO.abundance_unstratified.tsv [GO数据库 GO 丰度表] │ ├── All.GO.category.txt [GO数据库 GO 功能分类 丰度表] │ └── All.GO.detail.txt [GO数据库 GO 详细注释(最后几列) 丰度表] ├── 5-EC │ ├── All.LEVEL4EC.abundance_stratified.tsv [ENZYME数据库 酶 物种来源分层 丰度表] │ ├── All.LEVEL4EC.abundance.tsv [ENZYME数据库 酶 混合分层 丰度表, 相当于stratified+unstratified] │ └── All.LEVEL4EC.abundance_unstratified.tsv [ENZYME数据库 酶 丰度表] └── 6-CAZy ├── All.CAZY.abundance_stratified.tsv [CAZy数据库 碳水化合物活性酶 物种来源分层 丰度表] ├── All.CAZY.abundance.tsv [CAZy数据库 碳水化合物活性酶 混合分层 丰度表, 相当于stratified+unstratified] ├── All.CAZY.abundance_unstratified.tsv [CAZy数据库 碳水化合物活性酶 丰度表] ├── All.CAZY.Class.txt [CAZy数据库 碳水化合物活性酶 功能分类 丰度表] └── All.CAZY.detail.txt [CAZy数据库 碳水化合物活性酶 详细注释(最后几列) 丰度表]
3-AMRAnalysis/ ├── All.AMR.abundance_unstratified.tsv [CARD数据库 抗性基因 丰度表] ├── All.AMR.detail.txt [CARD数据库 抗性基因 详细注释(最后几列) 丰度表] ├── All.CARD.AMR.Gene.Family.txt [CARD数据库 抗性基因 基因家族 丰度表] ├── All.CARD.ARO.Name.txt [CARD数据库 抗性基因 基因名 丰度表] ├── All.CARD.Drug.Class.txt [CARD数据库 抗性基因 抗药性分类 丰度表] ├── All.CARD.Pathogen.txt [CARD数据库 抗性基因 病原分类 丰度表] ├── All.CARD.Resistance.Mechanism.txt [CARD数据库 抗性基因 抗性机制 丰度表] └── All.CARD.Short.Name.txt [CARD数据库 抗性基因 缩写名称 丰度表]
KEGG(KO),MetaCyc,EggNOG,GO,EC,CAZy的注释步骤
1)使用HUMAnN3软件[1] (HUMAnN2于2018年发表在Nature methods,随后维护升级至HUMAnN3), 将质控和去宿主之后的序列与蛋白质数据库(UniProt数据库UniRef90)进行比对(基于DIAMOND)
2)过滤掉比对失败的reads(HUMAnN3默认比对参数:translated_query_coverage_threshold = 90.0, prescreen_threshold = 0.01, evalue_threshold = 1.0, translated_subject_coverage_threshold = 50.0);
3)统计UniRef90各个蛋白的相对丰度(类似TPM, All.UniRef90.genefamilies.tsv)。 HUMAnN官方对原始丰度的解释如下:Gene family abundance is reported in RPK (reads per kilobase). This is computed as the sum of the scores for all alignments for a gene family. An alignment score is based on the number of matches to the reference gene for a specific sequence. It is divided by the length of the reference gene in kilobases to normalize for gene length. Each alignment score is also normalized to account for alignments for a single sequence to multiple reference genes. Alignments are not considered if they do not pass the e-value, identity, and coverage thresholds。 我们同时用HUMAnN提供的脚本humann_renorm_table 根据原始丰度计算相对丰度 (参数为:--units cpm,即copy per million, HUMAnN对此操作的描述是, CPM as used here does not refer to unnormalized COUNTS per million, but rather copies per million. CPMs as used here are a generic analog of the TPM (transcript per million) unit in RNA-seq. You may wish to use the abbreviation CoPM for added clarity)。 原文见https://github.com/biobakery/humann [1]。
4)功能和序列是多对多的关系 (一条序列可能对应多个功能,一个功能也可能对应多条序列), 无法直接计算功能的TPM丰度(无法校正序列长度), UniRef90和序列是一对一的关系, UniRef90可以理解为广义上的基因。 根据UniRef90 的ID 和各个功能数据库(KEGG, MetaCyc, EggNOG, GO, EC, CAZy)ID的对应关系(这个对应关系主要由 HUMAnN3 维护),累加属于同一个功能的基因(UniRef90)丰度, 得到各个功能数据库对应功能的相对丰度。 [1]
5)基于以上原理,不难发现基因(UniRef90)丰度表中,每个样本的总丰度是1 000 000。 而功能丰度表中,每个样本的总丰度可能就不是1 000 000。 这是因为功能丰度是由基因丰度计算的, 而功能和基因是多对多关系, 功能丰度表中, 一个基因的丰度可能被加零次或多次, 导致最终的总丰度 不等于1 000 000。
抗性基因(AMR)注释步骤
1)使用DIAMOND[2]软件 将各样本质控和去宿主之后的序列与CARD数据库进行比对, 过滤掉比对失败的序列;参数:-e 0.001 (e-value < 1e-3) --id 80 (percent identity > 80 %);
2)根据比对结果,统计出每个样本比对到各ARG参考序列的Fragments Count; 一对reads是按照一个整体(Fragment)来比对计数的, 也就是无论是一对reads的其中一条比对上,还是两条都比对上(同一个基因), 甚至是多部分比对上(考虑内含子), Fragments Count都只加1;RPKM/FPKM曾被许多高分文章广泛使用, 但最近TPM被认为是更好的基因丰度的计算方式, 因此我们根据Fragments Count计算TPM: TPM=(Fragments Count/Gene Length)/Sum(Fragments Count/Gene Length) [3];
3)同样,由于少部分ARO 与基因(数据库中的序列)存在多对多的关系, 累加之后, ARO丰度表中,每个样本的总丰度可能不等于1 000 000
KEGG数据库(KO)基本介绍:
KEGG 数据库于 1995 年由 Kanehisa Laboratories 推出 0.1 版,目前发展为一个综合性数据库,其中最核心的为 KEGG PATHWAY 和 KEGG ORTHOLOGY 数据库。在 KEGG ORTHOLOGY 数据库中,将行使相同功能的基因聚在一起,称为 Ortholog Groups (KO entries),每个 KO 包含多个基因信息,并在一至多个 pathway 中发挥作用。而在 KEGG PATHWAY 数据库中,将生物代谢通路划分为 6 类,分别为:细胞过程(Cellular Processes)、环境信息处理(Environmental Information Processing)、遗传信息处理(Genetic Information Processing)、人类疾病(Human Diseases)、新陈代谢(Metabolism)、生物体系统(Organismal Systems),其中每类又被系统分类为二、三、四层。第二层目前包括有 57个种子 pathway;第三层即为其代谢通路图;第四层为每个代谢通路图的具体注释信息。 >
MetaCyc数据库基本介绍
MetaCyc数据库是一个阐明通过实验手段阐释代谢通路的数据库。MetaCyc目标是收集所有已知生命的代谢通路,是一个庞大而全面的数据库,目前包含了来自3009个不同生物的2722个代谢通路。MetaCyc的代谢网络包含了初生代谢,次生代谢,还包括相关的化合物、酶和基因。
EggNOG数据库基本介绍
EggNOG数据库收集了COG(Clusters of Orthologous Groups of proteins,直系同源蛋白簇),构成每个COG的蛋白都是被假定为来自于一个祖先蛋白,因此是orthologs或者是paralogs。通过把所有完整基因组的编码蛋白一个一个的互相比较确定的。在考虑来自一个给定基因组的蛋白时,这种比较将给出每个其他基因组的一个最相似的蛋白(因此需要用完整的基因组来定义COG),这些基因的每一个都轮番地被考虑。如果在这些蛋白(或子集)之间一个相互的最佳匹配关系被发现,那么那些相互的最佳匹配将形成一个COG。这样,一个COG中的成员将与这个COG中的其他成员比起被比较的基因组中的其他蛋白更相像。
GO数据库基本介绍
GO(gene ontology)数据库是基因本体联合会(Gene Onotology Consortium)所建立的数据库,旨在建立一个适用于各种物种的,对基因和蛋白质功能进行限定和描述的,并能随着研究不断深入而更新的语言词汇标准。GO是多种生物本体语言中的一种,GO数据库根据功能将基因分为三大类:分子功能(MF),生物学过程(BP),细胞组分(CC)。
EC酶库基本介绍
ENZYME收录了酶的四级分类信息。EC编号或EC号是酶学委员会(Enzyme Commission)为酶所制作的一套编号分类法,每一个酶的编号都以字母“EC”起头,接着以四个号码来表示,这些号码代表逐步更细致的为酶作出分类。就如三肽胺基 蛋白酶的编号为EC3.4.11.4,当中的“EC3”是指水解酶(即以水来将分子分解的酶);“EC3.4”是那些与肽键产生作用的水解酶;“EC3.4.11”是单指那些从多胜肽中分开胺基末端的水解酶;“EC3.4.11.4”则是从三肽中分开胺基末端的水解酶。
CAZy数据库基本介绍
碳水化合物活性酶数据库(CAZy)是关于能够合成或者分解复杂碳水化合物和糖复合物的酶类的一个数据库资源,其基于蛋白质结构域中的氨基酸序列相似性,将碳水化合物活性酶类归入不同蛋白质家族。CAZy数据库提供了酶分子序列的家族信息,物种来源,基因序列,蛋白序列,三维结构,EC分类,相关数据库链接,建立此数据库是为了将酶分子的序列、结构、催化机制相关联。
碳水化合物活性酶数据库中基因ID前缀与功能分类的对应关系如下:
| 名称 | 缩写 |
|---|---|
| 糖苷水解酶类 | GHs |
| 糖苷转移酶类 | GTs |
| 多糖裂解酶类 | PLs |
| 糖水化合物脂酶类 | CEs |
| 碳水化合物结合模块 | CBMs |
| 辅助模块酶类 | AAs |
CARD数据库(抗性基因)基本介绍
细菌抗生素耐药性是在人类为控制细菌生长而进行的大规模抗生素的使用的前提下, 细菌通过基因突变或者从环境有机体的基因水平转移而获得对特定抗生素的抗性,这些突变使抗生素作用靶位发生改变或不能再对其靶标施加作用。耐药性的获得通常涉及到基因/移动DNA元件及其细菌宿主之间复杂的相互作用。随着DNA测序成本的降低和分析速度的提高, 研究者能够获得大量病原体基因组以及土壤、海洋和人类相关宏基因组的数据信息。利用基因组序列数据与生物信息学方法, 可深度剖析细菌耐药机制, 确定新药靶点, 寻找新的抗生素。CARD数据库目前使用最广泛的抗性基因(AMR)数据库,目前包括3997个抗性基因分类,并在线提供各个分类名称与PDB、NCBI等数据库的搜索接口,方便后续分析。
3.5 功能可视化( 2-FuctionAnalysis, 3-AMRAnalysis )
2-FuctionAnalysis/*-[功能数据库简写] 或 3-AMRAnalysis ├── 1-Barplots │ ├── [功能数据库简写]_[分组方案名称]_barplot.svg [总丰度前20功能丰度百分比堆积柱形图] │ ├── [功能数据库简写]_[分组方案名称]_groupMean_barplot.svg [总丰度前20功能分组平均丰度百分比堆积柱形图] │ ├── [功能数据库简写]_[分组方案名称]_groupMean_table.xls [总丰度前20功能分组平均丰度百分比统计表] │ └── [功能数据库简写]_[分组方案名称]_table.xls [总丰度前20功能丰度百分比统计表] ├── 2-Heatmaps │ ├── [功能数据库简写]_[分组方案名称]_clustered_heatmap.svg [总丰度前20功能log10(绝对丰度+1)聚类热图] │ ├── [功能数据库简写]_[分组方案名称]_clustered_table.xls [总丰度前20功能log10(绝对丰度+1)统计表] │ ├── [功能数据库简写]_[分组方案名称]_groupMean_heatmap.svg [总丰度前20功能分组平均(log10(绝对丰度+1))热图] │ ├── [功能数据库简写]_[分组方案名称]_groupMean_table.xls [总丰度前20功能分组平均(log10(绝对丰度+1))统计表] │ ├── [功能数据库简写]_[分组方案名称]_nocluster_heatmap.svg [总丰度前20功能log10(绝对丰度+1)热图] │ └── [功能数据库简写]_[分组方案名称]_nocluster_table.xls [总丰度前20功能log10(绝对丰度+1)统计表] ├── 3-Circos │ ├── [功能数据库简写]_[分组方案名称]_circos.png [样本功能丰度圈图, png格式] │ ├── [功能数据库简写]_[分组方案名称]_circos.svg [样本功能丰度圈图, svg格式] │ └── circos_conf [圈图数据文件] ├── 4-SignificanceAnalysis │ ├── DunnTest │ │ ├── [功能数据库简写]_DunnTest.xls [Kruskal-Wallis和Dunn’s Test检验结果表格] │ │ └── [功能数据库简写]_relative_abundance.xls [Kruskal-Wallis和Dunn’s Test检验输入表格] │ └── LEfSe │ ├── [功能数据库简写]_[分组方案名称]_lefse_LDA2.LDA.txt [LDA>2的LEfSe结果表] │ ├── [功能数据库简写]_[分组方案名称]_lefse_LDA2.lefseinput.txt [LEfSe对输入丰度表进行标准化后的丰度] │ ├── [功能数据库简写]_[分组方案名称]_lefse_LDA2.svg [LDA>2的LEfSe结果柱形图] │ ├── [功能数据库简写]_[分组方案名称]_lefse_LDA4.LDA.txt [LDA>4的LEfSe结果表] │ ├── [功能数据库简写]_[分组方案名称]_lefse_LDA4.svg [LDA>4的LEfSe结果柱形图] │ ├── [功能数据库简写]_[分组方案名称]_lefse.txt [LEfSe分析输入表] │ └── SignificantFeatures │ ├── [功能数据库简写]_[功能名称]_stratification_bar.svg [LEfSe LDA>2差异功能物种来源分层柱形图] │ └── table_for_stratification_bar.txt [差异功能物种来源分层柱形图输入表] ├── 5-CorrelationAnalysis │ ├── CorrelationHeatmap │ │ ├── [功能数据库简写]_Correlation_heatmap.svg [相关性热图] │ │ ├── [功能数据库简写]_p_value_matrix.xls [相关系数p值矩阵] │ │ └── [功能数据库简写]_spearman_rank_correlation_matrix.xls [spearman相关系数矩阵] │ └── RDA │ ├── [功能数据库简写]_[分组方案名称]_RDA.envfit.xls [RDA各环境因子坐标,解释方差,p值] │ ├── [功能数据库简写]_[分组方案名称]_RDA.Factors.PERMANOVA.xls [所有环境因子对样品功能丰度变异的解释方差的p值计算结果] │ ├── [功能数据库简写]_[分组方案名称]_RDA_features_location_plot.svg [RDA功能features点图] │ ├── [功能数据库简写]_[分组方案名称]_RDA.features.xls [RDA功能features点坐标] │ ├── [功能数据库简写]_[分组方案名称]_RDA.Group.PERMANOVA.xls [分组方式对样品功能丰度变异的解释方差的p值计算结果] │ ├── [功能数据库简写]_[分组方案名称]_RDA_sample_location_plot.svg [RDA样本点图] │ ├── [功能数据库简写]_[分组方案名称]_RDA_sample_location_plot_with_labels.svg[RDA样本点图(带样本名)] │ ├── [功能数据库简写]_[分组方案名称]_RDA.sample.xls [RDA样本点坐标] │ └── [功能数据库简写]_DCA_output.xls [DCA分析各排序轴值] └── * [功能丰度表见功能注释部分] 2-FuctionAnalysis/1-KEGG/ ├── 1-Barplots [同上] ├── 2-Heatmaps [同上] ├── 3-Circos [同上] ├── 4-SignificanceAnalysis [同上] ├── 5-ColoredMaps │ └── [KEGG 通路一级分类] │ └── [KEGG 通路二级分类] │ └── [KEGG 通路三级分类] │ ├── [KEGG 通路ID].html [KEGG 通路图网页版,标注LEfSe LDA>2 KO节点] │ └── [KEGG 通路ID].png [KEGG 通路图,标注LEfSe LDA>2 KO节点] └── 6-CorrelationAnalysis [同上5-CorrelationAnalysis]
3.5.1 功能统计 (1-Barplots, 2-Heatmaps, 3-Circos)
柱形图举例
根据数据库的注释结果, 绘制了各样品在各个功能层级上的相对丰度统计图(图3-5-1, 相对丰度前20)。

图3-5-1 KEGG代谢通路的Level1层级丰度柱形图
说明:不同的颜色代表不同的KEGG Pathway分类。
热图举例
从COG的相对丰度表出发,筛选出最大丰度排名前20的COG, 进行聚类和热图展示:

图3-5-2 各个样品COG聚类热图(相对丰度前20的基因)
说明:竖轴代表样品/分组信息,基于欧式距离聚类, 横轴的COG基于欧式距离聚类。
圈图举例
Circos 图可以用于展示每个样本各个功能(丰度前10)的比例 (如果整个丰度表最大丰度小于1,将等比例扩大1 000 000倍), 以及每个功能在各个样本中的比例:

图3-5-3 抗性基因在各个样本中的分布Circos 图
说明:左半圈为丰度最高的十个抗性基因, 每个抗性基因内,不同颜色代表不同样本来源的比例; 右边半圈为样本,样本内不同颜色代表不同抗性基因的比例。
3.5.2 功能差异(4-SignificanceAnalysis)
Kruskal-Wallis和Dunn’s Test检验
使用R语言dunn.test包[8] 进行Kruskal-Wallis和Dunn’s Test检验。 Kruskal-Wallis和Dunn’s Test都是基于秩次的非参数检验,对数据分布没有要求。 Kruskal-Wallis 秩和检验通常只能检验一个分组方案所有分组的整体差异, p<0.05只能推断至少有两个分组不一样,如果分组个数大于2, 那具体哪两个分组不一样是无法推断的。这时候我们需要结合Dunn’s Test来进行 多重比较,进而推断具体哪两个分组不一样。
Dunn’s Test的结果表格(DunnTest.xls)各列解释如下:
- Feature: feature标识
- KW_pvalue: Kruskal-Wallis秩和检验p值
- DunnTest_comparison: 分组对
- DunnTest_Z: 分组对Dunn’s Test检验统计量
- DunnTest_PValue: 分组对Dunn’s Test检验p值
- DunnTest_PValueBonferroniAdjusted: 分组对Dunn’s Test检验Bonferroni校正后的p值
LEfSe
LEfSe[9] (Linear discriminant analysis Effect Size,线性判别分析)即 LDA Effect Size 分析, 是一种发现和解释高纬度数据生物标识(分类单元、通路、基因)的分析工具,可以实现两 个或者多个分组之间的比较,同时也可进行分组内部亚组之间的比较分析。该分析首先使用非参数 Kruskal-Wallis 秩和 检测不同分组间丰度差异显著的功能 ,然后使用 Wilcoxon 秩和检验上一步的差异功能 在不同组间子分组中的差异一致性,最后采用线性回归分析(LDA)来估算每个功能 的丰度对差异效果影响的大小。 LEfSe寻找每一个分组的特征功能(默认为LDA>2的功能), 也就是相对于其他分组,在这个组中丰度较高的功能。 需要注意的是,LEfSe内部会对丰度进行标准化(主要计算每个样本参与分析功能的丰度比例), 特征功能原始丰度少数情况下不是最高的。

图3-5-4 KEGG数据库KO LEfSe分析LDA柱形图
每一横向柱形体代表一个功能,柱形体的长度对应LDA值,LDA值越高则差异越大。 柱形的颜色对应该功能是哪个分组的特征功能(在对应分组中的丰度相对较高)。
*.LDA.xls表格文件从左到右各列解释为:
- feature标识
- 所有样本标准化(算比例)以及log转化后的丰度均值
- 特征feature对应分组,如果为空,表示该feature不是特征feature
- LDA值
- Kruskal-Wallis 秩和检验p值
差异功能物种来源分层柱形图
这里需要特别说明的是,在LEfSe分析之后,我们挑出存在差异的功能(LDA>2), 用物种来源分层柱形图展示差异功能的物种来源,如3-5-5

3-5-5 KEGG数据库差异功能物种来源分层柱形图
KEGG 通路图(ColoredMaps)
根据KO (KEGG Orthologous groups) LEfSe组间差异比较结果, 我们可以进一步给map(通路)图上色, 在通路图中标注出检测到的基因,以及各个分组的特征基因(gene biomarkers) (图3-5-6)。
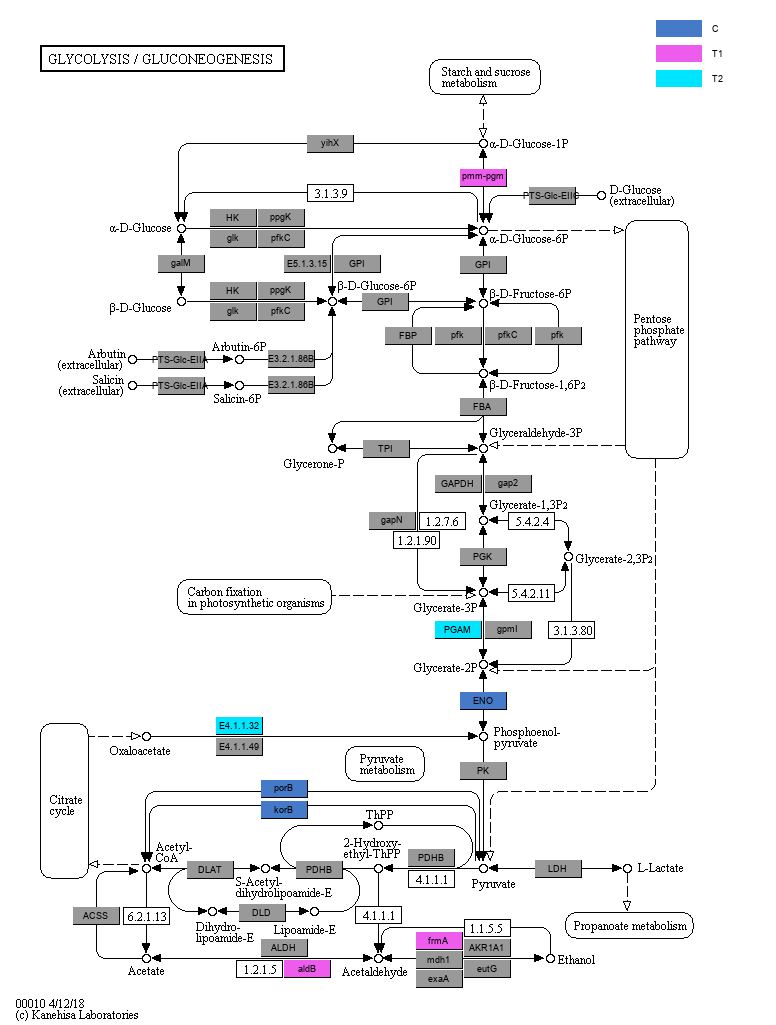
图3-5-6 通路中各个分组的biomarkers基因
说明:灰色矩形框表示在样本中检测到了该基因, 彩色矩形框是颜色对应分组的特征基因。同时请注意, 在本分析结果中,所有分组对应的颜色都是统一的。
另外,您可以打开同一文件夹下的同名网页文件(后缀不同), 查看每个通路矩形节点的详细信息, 鼠标悬在矩形框上可以看到您样本中检测到对应通路节点的哪些KOs, 以及这些KO是哪个分组的特征KO(KO后面括号内的分组, 表示这个KO是这个分组的特征KO), 点击矩形框可以查看KEGG官网对相应节点KOs的解释。 如下嵌入网页所示:
图3-5-6补充 通路中各个分组的biomarkers基因网页版
说明:尝试把鼠标悬在矩形框上,或者点击矩形框
3.5.3 功能相关性(CorrelationAnalysis)
此部分图与3.3.4小节的图一样, 参考3.3.4小节解读即可, 3.3.4小节里提到的微生物、物种, 相当于这里的功能
四 分析所用软件的版本
| 软件 | 版本 |
|---|---|
| HUMAnN3 | 3.6 |
| diamond | 0.9.14 |
| Kraken | 2.0.7-beta |
| Bracken | 2.0 |
| Trimmomatic | 0.39 |
| Bowtie2 | 2.3.5.1 |
| dunn.test | 1.3.5 |
| LEfSe | 1.0.8 |
| pheatmap | 1.0.12 |
| vegan | 2.6.0 |
| ggplot2 | 3.3.5.9000 |