微科盟16S扩增子数据分析结题报告
一 概述
微生物世界是分子多样性最大的天然资源库, 基于菌株水平的传统分离培养技术为人们认识微生物多样性提供了可能, 但是据估计自然界中超过99%的微生物不能通过传统的分离培养技术获得其纯培养, 从而导致环境微生物中的多样性基因资源难以被发现。 许多重要的微生物我们还不能识别,随着微生物活性产物的广泛研究和深入开发利用, 从环境微生物中筛选到新活性物质的几率将逐步下降。 而如何开拓利用环境微生物新资源是微生物研究的重要课题。 为此研究者们开发了多种以特定环境微生物为研究对象的高通量测序方法。
16S rRNA 测序技术是最常用的高通量测序依赖的组学技术之一, 该技术着眼于对肠道微生物群落菌种组成的分析。细菌16S rRNA基因具有保守区与可变区间隔排列的特征, 其中的可变区一般具有菌种特异性,并且可以反映细菌间亲缘关系的远近, 因此通过分析可变区的序列即可得到各细菌的分类学特征。 16S rRNA 基因序列包括9个可变区和10个保守区。 传统分子生物学方法中有时也应用 Sanger法对单一菌种的16S rRNA进行测序和鉴定, 而 16S rRNA 测序技术通过结合高通量测序技术的高通量优势和16S rRNA基因的菌种鉴定优势, 实现了对复杂样品中混合菌种的分类学鉴定和精确定量。 16SrRNA 测序技术的基本流程通过提取实验样品的DNA,并扩增16S rDNA某个可变区, 采用高通量测序仪Miseq或Hiseq对其进行测序, 通过生物信息学分析可以获得特定实验样品中细菌或古菌物种组成、物种丰度、系统进化、群落比较等诸多信息。
二 项目流程
2.1 测序实验流程
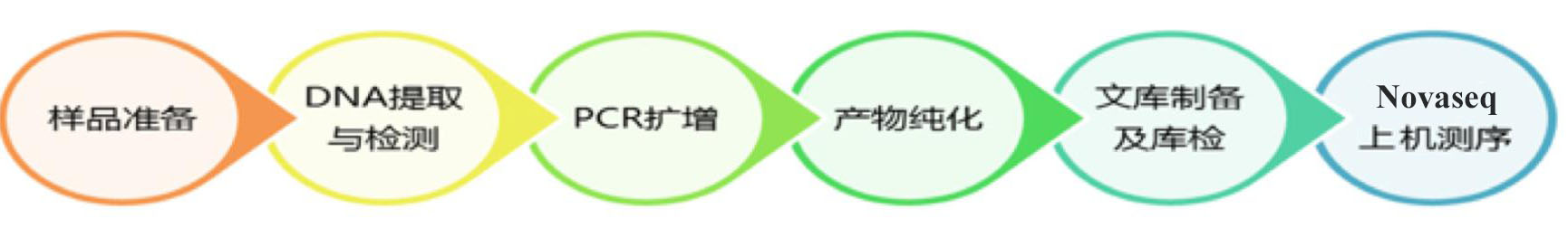
图2-1 实验工作流程图
2.2 生信分析流程
为了保证OTU(Operational Taxonomic Units)聚类及后续分析的准确性, 首先会对原始测序数据进行过滤处理,处理后的数据进行过滤得到有效数据。 然后基于有效数据进行OTU聚类/去噪和物种分类分析,形成OTU和其他物种分类等级的物种丰度谱。 基于数据均一化后的OTU物种丰度谱,再对OTU进行丰度、多样性指数等分析, 并对物种注释在各个分类水平上进行群落结构的统计分析。 还可以在以上分析的基础上,进行一系列的基于OTU、物种组成的聚类分析和统计比较分析, 挖掘样品之间的物种组成差异,结合环境及临床因子等进行关联统计分析, 寻找与其显著相关的物种群落,进行微生物群落的功能预测分析(图2-2)。
需要注意的是,生物信息分析流程分析使用的是QIIME2推荐的DADA2方法来去噪去嵌合, 相较QIIME1的UPARSE等聚类方法得出来的结果,DADA2比上一代分析的结果更准确。 因为不再按相似度聚类,所以生成的代表序列不再是OTU。 更准确的来说,在本报告中提到OTU应被称为扩增特征序列(ASV:Amplicon Sequence Variant或者Feature sequence)。 但为了方便理解,我们在报告中将以OTU来代表这个扩增特征的代表序列。 这也是目前研究扩增子领域研究人员通用的做法。
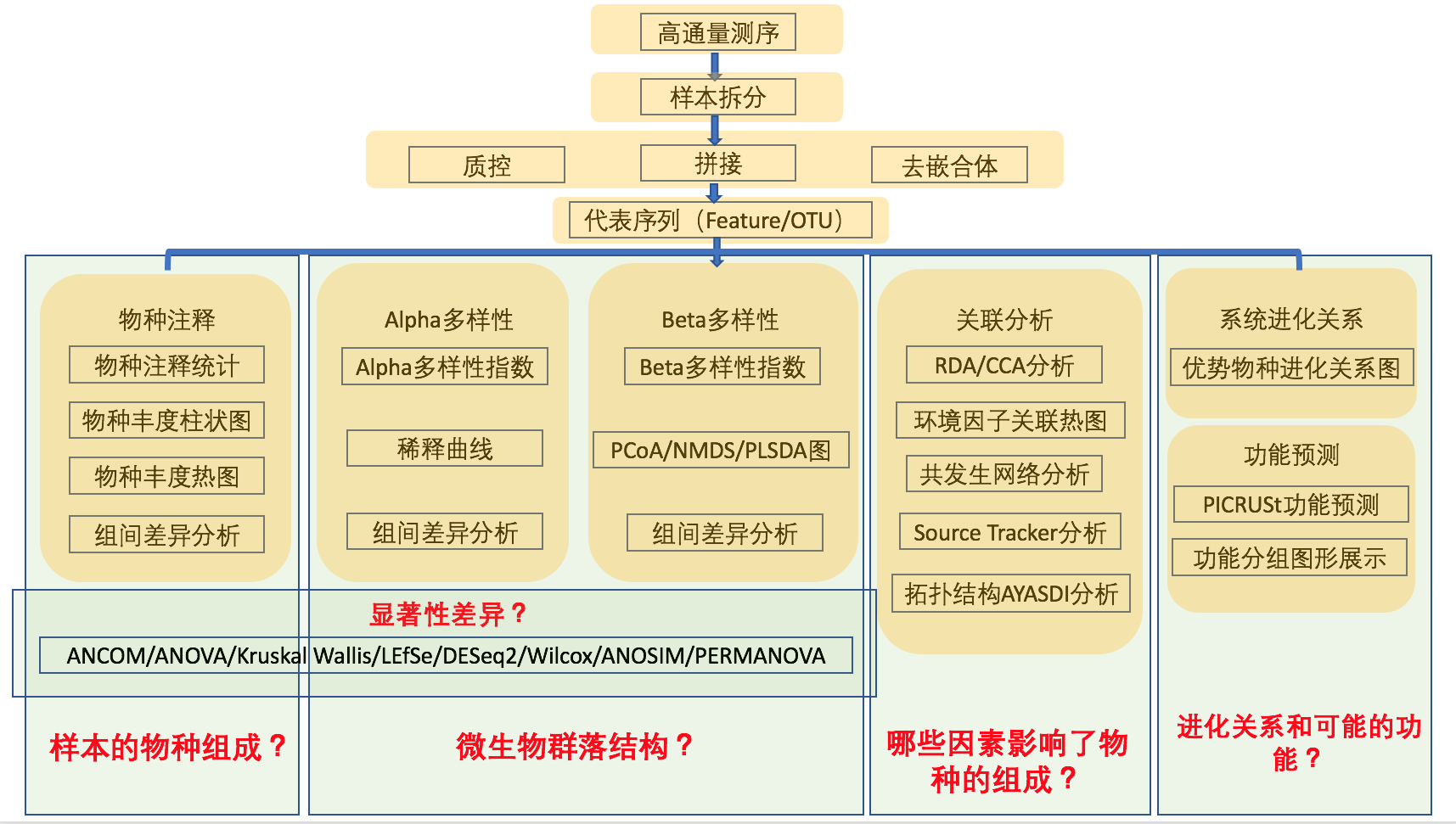
图2-2 生信分析流程图
三 结果文件夹解读
3.1 测序数据预处理
../1-OTUStats/ ├── 1-Stats-demux │ └── Summary_请点此文件查看.html [原始数据质量统计报告, 文件夹内其它文件皆为该报告的数据文件, 无需关注] ├── 2-Stats-OTU │ └── Summary_请点此文件查看.html [质控, 去噪, 拼接, 去嵌合体生成OTU过程的统计报告] ├── 3-RepresentiveSequence │ ├── dna-sequences.fasta [OTU代表序列] │ ├── rep-seqs │ │ └── Summary_请点此文件查看.html [OTU代表序列基本信息统计报告] │ ├── tree.rooted.nwk [有根进化树] │ └── tree.unrooted.nwk [无根进化树] ├── classified_stat_relative.csv [样本reads各水平物种注释比例统计表] ├── Classified_stat_relative.svg [样本reads各水平物种注释比例统计图] ├── feature-table.taxonomy.even.xls [OTU抽平丰度表] ├── feature-table.taxonomy.xls [OTU绝对丰度表] ├── number_of_assigned_otus.csv [样本各水平物种注释“OTU数量”统计表] ├── number_of_assigned_otus.svg [样本各水平物种注释“OTU数量”统计图] ├── number_of_taxa.csv [样本各水平物种注释“物种数量”统计表] └── number_of_taxa.svg [样本各水平物种注释“物种数量”统计图]
基于FASTQ格式的测序文件是一种存储序列信息的特定文件, 推荐用Notepad++等文本编辑器打开,每四行对应一条测序Read: 第一行以符号“@”起始,对应于序列ID和相应的描述信息; 第二行为实际测得的碱基序列;第三行以符号“+”起始; 而第四行的字符串则记录了第二行序列中每个碱基所对应的测序质量。
在本实验中得到的测序数据经过barcode拆分后获得的有效序列信息统计,如图3-1-1所示:
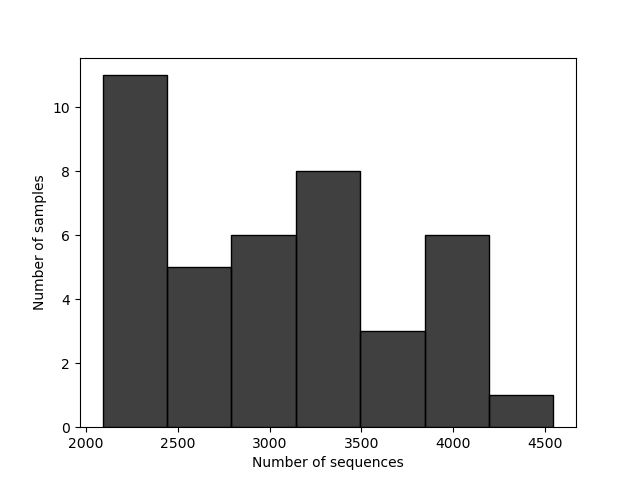
图3-1-1 数据拆分序列数目统计
说明:在数据按照barcode拆分之后每个样品的实际所含的序列(正向)数量统计。 X轴代表样品所含序列数目的区间,Y轴代表在这个所含序列数目的区间有多少个样品。 点击此处查看具体的样本测序量统计及序列测序质量统计结果(推荐)。
使用Qiime2软件 [1] 中的DADA2插件 [2] (云流程可选Deblur) 对所有样品的全部原始序列(input)进行质量控制(filtered), 去噪(纠正测序错误的序列,denoised),拼接(merged,单端数据则跳过), 并且去嵌合体(non-chimeric),形成OTU,表3-1-1显示了该过程各个步骤之后剩余序列数目。 DADA2/Deblur每一步的结果统计信息可以通过 点击此处打开新窗口查看 。
表3-1-1 dada2/Deblur去噪生成OTU的过程统计表格
| Unnamed: 0 | sample-id | reads-raw | fraction-artifact-with-minsize | fraction-artifact | fraction-missed-reference | unique-reads-derep | reads-derep | unique-reads-deblur | reads-deblur | unique-reads-hit-artifact | reads-hit-artifact | unique-reads-chimeric | reads-chimeric | unique-reads-hit-reference | reads-hit-reference | unique-reads-missed-reference | reads-missed-reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | YUN3259.2 | 2806 | 0.004633 | 0.000000 | 0.0 | 2451 | 2793 | 2020 | 2312 | 0 | 0 | 13 | 13 | 136 | 363 | 0 | 0 |
| 1 | BAQ2687.3 | 2417 | 0.003310 | 0.000414 | 0.0 | 1991 | 2410 | 1431 | 1790 | 1 | 1 | 17 | 17 | 97 | 399 | 0 | 0 |
| 2 | BAQ2420.1.1 | 2155 | 0.003248 | 0.000000 | 0.0 | 1870 | 2148 | 1506 | 1742 | 0 | 0 | 5 | 5 | 68 | 235 | 0 | 0 |
| 3 | BAQ2420.1.2 | 1984 | 0.002520 | 0.000000 | 0.0 | 1649 | 1979 | 1237 | 1521 | 0 | 0 | 2 | 2 | 84 | 305 | 0 | 0 |
| 4 | YUN3856.1.3 | 2163 | 0.002312 | 0.000925 | 0.0 | 1678 | 2160 | 1075 | 1506 | 2 | 2 | 15 | 15 | 103 | 499 | 0 | 0 |
| 5 | BAQ4166.2 | 3661 | 0.002185 | 0.000000 | 0.0 | 3123 | 3653 | 2405 | 2849 | 0 | 0 | 38 | 39 | 150 | 496 | 0 | 0 |
| 6 | BAQ3473.2 | 2786 | 0.002154 | 0.000359 | 0.0 | 2482 | 2781 | 2048 | 2294 | 1 | 1 | 14 | 14 | 167 | 369 | 0 | 0 |
| 7 | BAQ3473.3 | 3741 | 0.002138 | 0.000000 | 0.0 | 2992 | 3733 | 1994 | 2579 | 0 | 0 | 65 | 65 | 116 | 593 | 0 | 0 |
| 8 | BAQ4166.3 | 3512 | 0.001993 | 0.000000 | 0.0 | 2991 | 3505 | 2299 | 2718 | 0 | 0 | 18 | 18 | 165 | 519 | 0 | 0 |
| 9 | BAQ2687.1 | 2516 | 0.001987 | 0.000795 | 0.0 | 2174 | 2513 | 1724 | 1983 | 2 | 2 | 16 | 16 | 156 | 373 | 0 | 0 |
| 10 | YUN3428.2 | 4144 | 0.001931 | 0.000241 | 0.0 | 3370 | 4137 | 2378 | 3008 | 1 | 1 | 17 | 17 | 213 | 759 | 0 | 0 |
| 11 | BAQ3473.1 | 3233 | 0.001856 | 0.000309 | 0.0 | 2787 | 3228 | 2245 | 2620 | 1 | 1 | 55 | 55 | 147 | 437 | 0 | 0 |
| 12 | YUN3856.2 | 2898 | 0.001725 | 0.000000 | 0.0 | 2412 | 2893 | 1797 | 2213 | 0 | 0 | 12 | 12 | 173 | 543 | 0 | 0 |
| 13 | YUN3259.1.2 | 2020 | 0.001485 | 0.000000 | 0.0 | 1583 | 2017 | 1034 | 1404 | 0 | 0 | 6 | 6 | 93 | 416 | 0 | 0 |
| 14 | YUN3346.3 | 2054 | 0.001461 | 0.000487 | 0.0 | 1615 | 2052 | 1097 | 1444 | 1 | 1 | 8 | 9 | 93 | 393 | 0 | 0 |
| 15 | BAQ4166.1.3 | 2860 | 0.001399 | 0.000000 | 0.0 | 2304 | 2856 | 1610 | 2065 | 0 | 0 | 27 | 27 | 127 | 516 | 0 | 0 |
| 16 | YUN3259.3 | 2940 | 0.001361 | 0.000000 | 0.0 | 2453 | 2936 | 1776 | 2124 | 0 | 0 | 12 | 12 | 118 | 411 | 0 | 0 |
| 17 | YUN3533.1.3 | 2261 | 0.001327 | 0.000000 | 0.0 | 1883 | 2258 | 1446 | 1763 | 0 | 0 | 21 | 21 | 155 | 434 | 0 | 0 |
| 18 | BAQ4697.3 | 3146 | 0.001271 | 0.000000 | 0.0 | 2283 | 3142 | 1307 | 1995 | 0 | 0 | 56 | 57 | 104 | 754 | 0 | 0 |
| 19 | BAQ4697.2 | 2374 | 0.001264 | 0.000000 | 0.0 | 1752 | 2371 | 980 | 1489 | 0 | 0 | 52 | 52 | 90 | 576 | 0 | 0 |
| 20 | YUN3428.3 | 3298 | 0.001213 | 0.000000 | 0.0 | 2813 | 3294 | 2156 | 2567 | 0 | 0 | 17 | 17 | 187 | 539 | 0 | 0 |
| 21 | BAQ2462.1 | 2553 | 0.001175 | 0.000000 | 0.0 | 2006 | 2550 | 1211 | 1659 | 0 | 0 | 17 | 18 | 79 | 473 | 0 | 0 |
| 22 | YUN3533.2 | 3433 | 0.001165 | 0.000291 | 0.0 | 2482 | 3430 | 1595 | 2314 | 1 | 1 | 30 | 32 | 168 | 858 | 0 | 0 |
| 23 | YUN2029.2 | 2626 | 0.001142 | 0.000000 | 0.0 | 1778 | 2623 | 705 | 1410 | 0 | 0 | 38 | 38 | 56 | 722 | 0 | 0 |
| 24 | BAQ2687.2 | 1830 | 0.001093 | 0.000000 | 0.0 | 1401 | 1828 | 741 | 1127 | 0 | 0 | 2 | 2 | 46 | 397 | 0 | 0 |
| 25 | YUN3533.3 | 3691 | 0.001084 | 0.000000 | 0.0 | 3012 | 3687 | 2189 | 2757 | 0 | 0 | 19 | 21 | 213 | 718 | 0 | 0 |
| 26 | YUN3856.1.1 | 2892 | 0.001037 | 0.000000 | 0.0 | 2506 | 2889 | 1981 | 2306 | 0 | 0 | 17 | 17 | 99 | 340 | 0 | 0 |
| 27 | YUN3428.1 | 2916 | 0.001029 | 0.000000 | 0.0 | 2502 | 2913 | 1893 | 2237 | 0 | 0 | 9 | 9 | 160 | 451 | 0 | 0 |
| 28 | YUN3346.1 | 1947 | 0.001027 | 0.000000 | 0.0 | 1427 | 1945 | 779 | 1217 | 0 | 0 | 5 | 5 | 87 | 482 | 0 | 0 |
| 29 | YUN1242.3 | 2123 | 0.000942 | 0.000000 | 0.0 | 1358 | 2121 | 471 | 1080 | 0 | 0 | 18 | 18 | 30 | 596 | 0 | 0 |
| 30 | YUN3856.3 | 3628 | 0.000827 | 0.000000 | 0.0 | 2962 | 3625 | 2128 | 2703 | 0 | 0 | 33 | 33 | 213 | 724 | 0 | 0 |
| 31 | BAQ4166.1.2 | 3692 | 0.000813 | 0.000000 | 0.0 | 3162 | 3689 | 2380 | 2795 | 0 | 0 | 29 | 30 | 175 | 546 | 0 | 0 |
| 32 | YUN3533.1.2 | 2719 | 0.000736 | 0.000000 | 0.0 | 2378 | 2717 | 1954 | 2240 | 0 | 0 | 5 | 5 | 195 | 425 | 0 | 0 |
| 33 | BAQ4166.1.1 | 2945 | 0.000679 | 0.000340 | 0.0 | 2613 | 2944 | 2159 | 2410 | 1 | 1 | 37 | 37 | 147 | 348 | 0 | 0 |
| 34 | BAQ2420.1.3 | 1952 | 0.000512 | 0.000000 | 0.0 | 1545 | 1951 | 1117 | 1457 | 0 | 0 | 1 | 1 | 79 | 341 | 0 | 0 |
| 35 | YUN1609.1 | 2054 | 0.000487 | 0.000000 | 0.0 | 1383 | 2053 | 453 | 973 | 0 | 0 | 2 | 2 | 30 | 532 | 0 | 0 |
| 36 | YUN1005.1.1 | 2429 | 0.000412 | 0.000000 | 0.0 | 1573 | 2428 | 550 | 1244 | 0 | 0 | 9 | 9 | 35 | 665 | 0 | 0 |
| 37 | YUN3856.1.2 | 2765 | 0.000362 | 0.000000 | 0.0 | 2088 | 2764 | 1359 | 1924 | 0 | 0 | 17 | 17 | 165 | 696 | 0 | 0 |
| 38 | YUN3533.1.1 | 2818 | 0.000355 | 0.000355 | 0.0 | 2308 | 2818 | 1667 | 2073 | 1 | 1 | 10 | 10 | 76 | 401 | 0 | 0 |
| 39 | BAQ4697.1 | 2642 | 0.000000 | 0.000000 | 0.0 | 1926 | 2642 | 995 | 1564 | 0 | 0 | 32 | 32 | 86 | 644 | 0 | 0 |
重要 : 除了通过网页版报告,用户也可以通过本地文件夹查看结果, 网页报告只选取了部分代表性信息用于引导用户阅读, 然而本地文件夹的结果目录中包含了大量其他有用的信息。 需要注意的是,结果文件夹中有一些特殊的文件夹, 这些文件夹中包含HTML文件“Summary_请点此文件查看.html”, 这类文件夹是qiime2 qzv文件解压缩后得到的, 直接打开网页“Summary_请点此文件查看.html”可查看所有有用信息, 不用理会文件夹中其它的网页脚本和数据文件。 “Summary_请点此文件查看.html”网页中的图片能够进行多种个性化的调整 (如颜色,背景,分组方式,视角等),建议好好探索和利用,以挖掘结果中的信息。
3.2 物种的注释与评估
2-AbundanceAnalysis ├── 1-AbundanceSummary │ ├── 1-AbundanceTable │ │ ├── 1-Absolute │ │ │ └── otu_table.[物种分类水平].absolute.xls [对应水平绝对丰度表] │ │ ├── 2-Relative │ │ │ └── otu_table.[物种分类水平].relative.xls [对应水平相对丰度表] │ │ ├── 3-RelativeUnspecified │ │ │ └── otu_table.[物种分类水平].relative.xls [相对丰度表,OTU在某个水平没有分类,则使用"Unspecified_上一级有分类的水平"代替] │ │ ├── 4-Even │ │ │ └── otu_table.[物种分类水平].even.xls [对应水平抽平丰度表] │ │ └── 5-CollapsedStats │ │ └── collapsed-[物种分类水平] │ │ └── Summary_请点此文件查看.html [对应水平物种分类情况统计] │ ├── 2-Barplots │ │ ├── taxa-bar-plots_Qiime2 │ │ │ └── Summary_请点此文件查看.html [Qiime2 物种 百分比(相对丰度)堆积柱形图, 可手动调整物种水平] │ │ ├── taxa-bar-plots-top20 │ │ │ ├── [物种分类水平]_[分组方案]_barplot.svg [对应水平总丰度前20物种丰度百分比堆积柱形图] │ │ │ └── [物种分类水平]_[分组方案]_table.xls [对应水平总丰度前20物种丰度百分比统计表] │ │ └── taxa-bar-plots-top20-group-mean │ │ ├── [分组方案]_[物种分类水平]_mean_barplot.svg [对应水平总丰度前20物种分组平均丰度百分比堆积柱形图] │ │ └── [分组方案]_[物种分类水平]_mean_table.xls [对应水平总丰度前20物种分组平均丰度百分比统计表] │ └── 3-Heatmaps │ ├── Heatmap_top20 │ │ └── [物种分类水平] │ │ ├── group_mean_heatmap.svg [对应水平总丰度前20物种分组平均(log10(绝对丰度+1))热图] │ │ ├── group_mean_table.xls [对应水平总丰度前20物种分组平均(log10(绝对丰度+1))统计表] │ │ ├── heatmap.svg [对应水平总丰度前20物种log10(绝对丰度+1)热图] │ │ └── table.xls [对应水平总丰度前20物种log10(绝对丰度+1)统计表] │ └── Heatmap_top20_clustered │ └── [物种分类水平] │ ├── heatmap.svg [对应水平总丰度前20物种log10(绝对丰度+1)聚类热图] │ └── table.xls [对应水平总丰度前20物种log10(绝对丰度+1)统计表] ├── 2-AbundanceComparison │ ├── 1-ANCOM │ │ └── [分组方案].ANCOM.[物种分类水平] │ │ └── Summary_请点此文件查看.html [ANCOM分析报告] │ ├── 2-ANOVA │ │ └── ANOVA_[分组方案]_DiffAbundance_[物种分类水平].xls [ANOVA分析结果表] │ ├── 3-KruskalWallis │ │ └── kruskal_wallis_[分组方案]_DiffAbundance_[物种分类水平].xls [KruskalWallis分析结果表] │ ├── 4-LEfSe │ │ └── [物种分类水平] │ │ ├── [分组方案]_[物种分类水平]_lefse_LDA2.cladogram.svg [LDA>2的LEfSe结果cladogram图] │ │ ├── [分组方案]_[物种分类水平]_lefse_LDA2.LDA.xls [LDA>2的LEfSe结果表] │ │ ├── [分组方案]_[物种分类水平]_lefse_LDA2.lefseinput.xls [LEfSe对输入丰度表进行标准化后的丰度] │ │ ├── [分组方案]_[物种分类水平]_lefse_LDA2.svg [LDA>2的LEfSe结果柱形图] │ │ ├── [分组方案]_[物种分类水平]_lefse_LDA4.cladogram.svg [LDA>4的LEfSe结果cladogram图] │ │ ├── [分组方案]_[物种分类水平]_lefse_LDA4.LDA.xls [LDA>4的LEfSe结果表] │ │ ├── [分组方案]_[物种分类水平]_lefse_LDA4.svg [LDA>4的LEfSe结果柱形图] │ │ └── [分组方案]_[物种分类水平]_lefse.xls [LEfSe分析输入表] │ └── 5-DESeq2 │ ├── [分组方案]_numerator_[处理组]_denominator_[对照组]_[分类水平]_DESeq2.svg [DESeq2结果图] │ └── [分组方案]_numerator_[处理组]_denominator_[对照组]_[分类水平]_DESeq2.xls [DESeq2结果表] ├── [分组方案]_classified_stat_relative.csv [分组方案对应样本reads各水平物种注释比例统计表] ├── [分组方案]_Classified_stat_relative.svg [分组方案对应样本reads各水平物种注释比例统计图] ├── [分组方案]_feature-table.taxonomy.even.xls [分组方案对应样本OTU抽平丰度表] ├── [分组方案]_feature-table.taxonomy.xls [分组方案对应样本OTU绝对丰度表] ├── [分组方案]_number_of_assigned_otus.csv [分组方案对应样本各水平物种注释“OTU数量”统计表] ├── [分组方案]_number_of_assigned_otus.svg [分组方案对应样本各水平物种注释“OTU数量”统计图] ├── [分组方案]_number_of_taxa.csv [分组方案对应样本各水平物种注释“物种数量”统计表] └── [分组方案]_number_of_taxa.svg [分组方案对应样本各水平物种注释“物种数量”统计图]
3.2.1 物种组成分析
为了研究样品物种组成及多样性信息, 对所有样品的全部的有效序列进行聚类/去噪,形成OTU,也可以称为特征序列。 选取OTU的代表性序列,与数据库 ( 16S 默认Greengenes2数据库[3], 云流程可选SILVA数据库[4] ) 进行比对获得物种注释信息(默认qiime feature-classifier classify-sklearn方法 [1] )。基于物种注释信息,去除注释为叶绿体、 线粒体以及不能注释到界级别的OTU及其包含的序列。基于OTU的绝对丰度及注释信息, 对每个样品在一共7个分类水平界门纲目科属种 (Kingdom,Phylum,Class,Order,Family,Genus,Species) 上的序列数目占总序列数的比例进行统计,可以有效地评估样本的物种注释分辨率 (注释到属/种的比例越高表示样本的OTU注释效果越好,但不表征注释的准确性), 图3-2-1展示了每个样本中OTU在各分类水平注释的相对程度:

图3-2-1 各个样品在各分类水平上的序列注释程度柱形图
说明:横坐标(Sample Name)是样品名, 纵坐标(Sequence Number Percent)表示注释到该水平的序列数目 占总注释数据的比率,柱状图自上而下的颜色顺序对应于右侧的图例颜色顺序。 每个分类水平最高值为1,代表100%的序列都得到了至少在这个级别的注释。
图3-2-2展示了样本中物种在不同分类水平相对分布情况的柱形图:
图3-2-2 各个样本在各分类水平的物种相对分布情况的柱形图
说明:横坐标(Sample)是样品名, 纵坐标(Relative Frequency)表示相对丰度。 其他分类水平的物种相对丰度图也都可以通过交互式网页打开, 并对样本或者OTU在图片中呈现的顺序根据元数据(包括分组信息) 或者相对丰度的大小进行调节。 Level 1,2,3,4,5,6,7依次代表分类水平界门纲目科属种。

图3-2-3 各个分组在门水平上的相对分布情况柱形图(相对丰度前20的物种)
说明:通常在种/属水平因为分类种类过多而在图例中无法全部展示所有分类, 特增加本图是对图3-2-2进行补充。横坐标是分组名, 纵坐标(Sequence Number Percent)表示注释到该门水平的序列数目 占总注释数据的比率,柱状图自上而下的颜色顺序对应于右侧的图例颜色顺序。 在门水平没有注释的序列被归为unclassified一类。 更多完整的物种分布情况请参见结果部分 2-AundanceAnalysis/1-AbundanceSummary/2-Barplots/ 。图例中最多显示最优势的20个种类, 余下的相对丰度较低的物种被归类为Other在图中展示。
为了研究不同样品间的相似性, 还可以通过对样品进行聚类分析从而构建样品的聚类树。 选取关注分类水平的物种(默认选取物种绝对丰度前20)实现样品聚类, 以此考察不同样品或者分组间的相似或差异性(此处为门水平分类的热图), 并从分类信息和样品间差异两个层面进行横向聚类, 寻找物种或样本的聚集规律。结果中也提供了不聚类样品, 按分组顺序排列的热图。

图3-2-4 门分类水平的热图
说明:纵向为样品名称信息。横向为门水平分类注释名称。 图中上方的聚类树为物种在所有样品中的丰度分布相似程度聚类, 左侧的聚类树为样品的物种丰度分布相似程度聚类。 中间的热图是log10(绝对丰度)热图, 其他分类等级完整的热图聚类结果请参见结果部分 2-AundanceAnalysis/1-AbundanceSummary/3-Heatmaps/ 。
3.2.2 组间OTU差异显著性分析
ANCOM(Analysis of composition of microbiomes) [5] 是专门用于比较微生物组学数据中物种在组间的显著性差异的分析方法。 ANCOM分析不依赖于数据的分布假设, 并解决了在其他方法中相对丰度分析所带来的限制, 从而能够有效降低结果的假阳性。 从下面链接中我们可以得知属水平分类的物种在组间表现了丰度的显著差异:
图3-2-5 属水平分类的ANCOM丰度比较结果
说明:在ANCOM分析中,W值是一个衡量组间差异显著性的统计量(类似F值,t值), W值越高,代表该物种在组间的差异显著性越高。 图中的每一个点都代表了一个比较的物种, 纵坐标代表W值,横坐标clr值代表组间样品丰度的差异程度, 数字越高代表相对丰度差异越大。因此在图中的点越靠近右上角, 则代表该物种与其他物种(大部分集中在左下角)相比更具有显著性差异。 组间具有差异显著的物种被列在下面相应的表格中。
同时Kruskal Wallis/ANOVA/LEfSe/DESeq2 等分析也被用于物种各分类水平在组间的显著性差异: Kruskal Wallis和ANOVA的分析基于均一化的丰度表, 运用了qiime1 group_significance.py脚本; ANCOM的分析基于绝对丰度表(所有值加1),运用了qiime2 composition ancom命令。 ANCOM、Kruskal Wallis和ANOVA用于多组间丰度差异的显著性检验。 Kruskal Wallis和ANOVA提供检验统计量(Test-Statistic),总的p值(P), 校正错误发现率p值(FDR_P),Bonferroni校正p值(Bonferroni_P), 及各分组均值,但不提供多重比较的结果;ANOVA是参数检验,要求丰度服从正态分布, 而大部分物种的丰度都是不服从正态分布的;Kruskal Wallis属于非参数检验, 对分布没有要求,较为适合菌群分析。LEfSe [6] 方法基于相对丰度表, 是非参数检验和线性判别分析的结合,适合菌群丰度差异检验; LEfSe寻找每一个分组的特征微生物(LDA>阈值的微生物,为了方便客户选择, 我们在结果文件夹中分别提供了阈值为2和4的结果),也就是相对于其他分组, 在这个组中丰度较高的微生物(如图3-2-6);同时, 图3-2-7标注了特征微生物的分类层级所属关系, 科以上有差异的大分类在图中标注分类区间和名字,科以下的只区分颜色, 由图可以推断在分组间有显著差异的微生物的进化关系; DESeq2运用了R语言DESeq2包, [7] 结果表格的文件名称格式为 {category}_numerator_{group1}_denominator_{group2}_{level}_DESeq2.txt, 其中baseMean表示所有样本的丰度平均值, log2FoldChange对应log2(group1/group2), 一般情况下log2FoldChange大于零代表group1的丰度大于group2的丰度, 也有相反的情况,这是由于DESeq2假设FoldChange服从正态分布 (相比group2,group1丰度高和丰度低的物种应该各占一半), 如果您样本中大部分物种都是group1偏多或者group2偏多, DESeq2校正(估计)FoldChange后就会出现相反的情况, lfcSE对应log2FoldChange的标准误,该值越小则说明FoldChange估计得越准确, stat对应检验统计量,pvalue 和padj分别对应p值和校正p值。 我们用DESeq2方法在分组间进行多重比较,寻找每两个分组之间显著差异的微生物。
这五种显著性差异比较方法各有优劣并且对结果可能会有不同的解释, 是属于合理的现象,用户可以根据数据本身的特性去选择方法解读数据。 DESeq2和ANCOM两种方法都是基于比例来进行差异分析的, 相较于传统的相对丰度,比例的好处在于,样本中某一个物种丰度发生变化, 在统计上并不会影响另一个物种的丰度,而相对丰度则不然。 我们推荐使用LEfSe先去查看某物种在多组之间的丰度差异, 再使用DESeq2去了解具体的两两分组比较结果。 完整的差异性分析结果请见结果部分 2-AundanceAnalysis/2-AbundanceComparison/ 。

图3-2-6 LEfSe分析LDA柱形图
说明:每一横向柱形体代表一个物种,柱形体的长度对应LDA值, LDA值越高则差异越大。柱形的颜色对应该物种是哪个分组的特征微生物, 特征微生物(Biomarkers)表示在对应分组中的丰度相对较高。

图3-2-7 LEfSe分析cladogram图
说明:cladogram图由内到外,分别对应界门纲目科属不同的分类层级, 层级间连线代表所属关系。每个圆圈节点代表一个物种, 节点为黄色代表在分组间差异不显著,不为黄色则代表该物种是对应颜色分组的特征微生物 (在该分组丰度显著较高)。有颜色的扇形区域标注了特征微生物的下属分类区间。
[分组方案]_[物种分类水平]_lefse_LDA2.LDA.xls表格文件从左到右各列解释为:
- feature标识
- 所有样本标准化(筛除无意义feature, 算相对丰度后乘以1000000) 以及log转化后的丰度均值
- 特征feature对应分组,如果为空,表示该feature不是特征feature
- LDA值
- Kruskal-Wallis 秩和检验p值
3.2.3 样品共有物种分析
对于样本间,根据OTU是否存在来寻找样本之间的特有或共有的OTU, 对于分组较少(小于等于5)的实验方案,我们绘制韦恩图 (Venn diagram)分析不同样品组之间特有或共有的OTU, 用于统计多个样本中所共有和独有的OTU数目, 可以比较直观的表现样本在OTU水平上的组成相似性及重叠情况(图3-2-8)。 对于分组较多的(大于等于3),我们绘制了花瓣图,花瓣图中, 花瓣里是对应分组特有的OTU数目,中心是所有分组共有的OTU数目。 同时也给出了特有和共有的OTU种类,注释情况及丰度, 丰度较高且特有的OTU具有重要分析价值, 请参见 1-VennAndFlower

查看各分类水平的物种在各个样本的分布情况请点击 2-AundanceAnalysis/1-AbundanceSummary/1-AbundanceTable/5-CollapsedStats/ 。
3.3 Alpha多样性分析
3-AlphaDiversity/
├── 1-AlphaDiversitySummary
│ ├── alpha-summary.xls [各样本所有多样性指数汇总表]
│ └── [分组方案名]_alpha_diversity_[α多样性指数名称].boxplot.svg [对应α多样性指数箱线图]
├── 2-AlphaRarefaction
│ ├── alpha-rarefaction-ggplot2
│ │ └── [α多样性指数名称]_rarefaction_curve.svg [对应α多样性指数“拟合”稀释性曲线]
│ └── alpha-rarefaction-Qiime2
│ └── Summary_请点此文件查看.html [各α多样性指数稀释性曲线网页报告(可在网页内选择指数)]
└── 3-SignificanceAnalysis
├── 1-Wilcox_Test
│ └── [α多样性指数名称]_[分组方案名]_wilcox_compare_boxplot.svg [对应α多样性指数wilcox多重比较箱线图]
└── 2-Kruskal_Wallis
└── [α多样性指数名称]-group-significance
└── Summary_请点此文件查看.html [对应α多样性指数Kruskal Wallis比较网页报告]
3.3.1 Alpha多样性指数统计分析
Alpha多样性以及Beta多样性的分析主要用qiime2 diversity插件 [1] 完成。Alpha多样性指数是对某个样品中物种多样性的分析, 包含样品中的物种组成的丰富度和均匀度两个因素, 通常用Observed OTUs(observed_features), Shannon以及Faith's Phylogenetic Diversity等指数 来评估某个样本的物种多样性,指数越高,表明样本的多样性越复杂。 Observed OTUs指数是指样本中实际测定得到的OTU数量, 衡量样品中OTU丰富度的指数。Shannon指数, 它的计算考虑到样品中的OTU总数,和每个OTU所占的比例。 Faith's Phylogenetic Diversity是基于系统发生树来计算的 一种多样性指数,它用各个样品中OTU的代表序列计算出构建系统发生树的距离, 将某一样品中的所有代表序列的值加和,从而得到的数值。 chao1指数是用来反映物种丰富度(物种的种类数量)的指标。 它通过观测到的结果推算出一个理论的丰富度, 这个丰富度更接近真实的丰富度。

图3-3-1 Shannon指数的箱型图
说明:不同颜色代表不同的分组。 其他Alpha多样性指数的箱型图请参见结果部分 3-AlphaDiversity/1-AlphaDiversitySummary/ 。
Alpha多样性稀释曲线(Rarefaction Curve)是 从样品中随机抽取一定测序量的数据, 统计它们所代表物种数目或多样性指数, 以测序数据量与物种多样性来构建的曲线, 以用来说明样品的测序数据量是否合理,并间接反映样品中物种的丰富程度。 曲线的平缓程度反映了测序深度对于观测样本多样性的影响大小, 曲线越平缓,表明测序结果已足够反映当前样本所包含的多样性, 继续增加测序深度已无法检测到大量的尚未发现的新OTU;反之, 则表明多样性尚未接近饱和,继续增加测序深度将有助于观测到更多的新OTU。
图3-3-2 各Alpha多样性指数的稀释性曲线
说明:通过选择下拉框选择不同的Alpha多样性指数或者分组情况 (网页上半部分图),图中X轴代表抽取的序列测序量, Y轴代表相应的Alpha多样性指数值。 网页下半部分图代表在相应抽取测序量时, 各分组中仍包含样品数量,用于查看在一定抽取测序量之下 稀释曲线的分组分析是否仍然包括该分组中全部的样本。
3.3.2 Alpha多样性指数组间差异检验
在得到整体的Alpha多样性指数之后, 接下来结合分组信息来用Kruskal Wallis方法比较 在各个样品分组之间Alpha多样性指数是否有显著性差异。 Alpha多样性指数Kruskal Wallis方法比较结果文件为 3-AlphaDiversity/3-SignificanceAnalysis/2-Kruskal_Wallis/ 目录下的相关文件。除了使用QIIME2推荐的Kruskal Wallis方法, 分析还包括使用Wilcox Test去精确比较了各个组间的显著性差异并做图, 图3-3-3显示了Shannon指数的组间多重比较结果。

图3-3-3 Shannon指数的组间多重比较
说明:X轴表示分组名称,Y轴表示Alpha多样性指数。 *,**,***分别代表p<0.05, p<0.01, p<0.001. 其他指数的完整信息请查看 3-AlphaDiversity/3-SignificanceAnalysis/1-Wilcox_Test
3.4 Beta多样性分析
4-BetaDiversity
├── 1-BetaDiversitySummary
│ ├── BetaDiversity_heatmap.svg [所有β多样性指数热图]
│ ├── unclustered_[β多样性指数名称]_betadiversity_summary.svg [对应β多样性指数非聚类热图]
│ ├── [β多样性指数名称]_betadiversity_summary.svg [对应β多样性指数聚类热图]
│ └── [β多样性指数名称]_matrix.xls [对应β多样性指数矩阵表]
├── 2-PCoA
│ ├── PCoA-Phyloseq
│ │ ├── [分组方案名]_[β多样性指数名称]_PCoA_2D.svg [2D PCoA图]
│ │ ├── [分组方案名]_[β多样性指数名称]_PCoA_2D_without_labels.svg [2D PCoA图, 不标注样本名]
│ │ ├── [分组方案名]_[β多样性指数名称]_PCoA_3D.svg [3D PCoA图]
│ │ └── [分组方案名]_[β多样性指数名称]_PCoA.ord.xls [PCoA各样本坐标]
│ └── PCoA-Qiime2
│ └── [β多样性指数名称]_emperor
│ └── Summary_请点此文件查看.html [对应β多样性指数QIIME PCoA网页报告, 可动态调整图片]
├── 3-NMDS
│ ├── [分组方案名]_[β多样性指数名称]_NMDS.ord.xls [NMDS各样本坐标表格]
│ ├── [分组方案名]_[β多样性指数名称]_NMDS.svg [NMDS图]
│ └── [分组方案名]_[β多样性指数名称]_NMDS_without_labels.svg [NMDS图, 不标注样本名]
├── 4-PLS-DA
│ ├── [分组方案名]_PLSDA_AUC_plot.svg [PLSDA ROC曲线]
│ ├── [分组方案名]_PLSDA_comp_plot.svg [PLSDA 坐标图]
│ └── [分组方案名] _ PLSDA_Variable_importance_in_projection.xls [PLSDA, 各OTU 的重要性(VIP), 看comp.1就行]
├── 5-GroupSignificance
│ ├── [β多样性指数名称]-anosim-[分组方案名]-significance
│ │ └── Summary_请点此文件查看.html [对应β多样性指数anosim分析报告]
│ └── [β多样性指数名称]-permanova-[分组方案名]-significance
│ └── Summary_请点此文件查看.html [对应β多样性指数permanova分析报告]
└── 6-PCA
├── [分组方案名]_PCA_plot.svg [PCA 图]
└── [分组方案名]_PCA_plot_without_lables.svg [PCA 图, 不标注样本名]
3.4.1 Beta多样性指数统计分析
Beta Diversity是对不同样品间的微生物群落构成进行比较。 根据样本的OTU丰度信息计算Bray Curtis,Weighted Unifrac 和Unweighted Unifrac距离来评估不同样品间的微生物群落结构差异。 Bray Curtis距离是生态学上反应群落之间差异性最常用的指标, 只考虑了物种的丰度信息。 Unweighted Unifrac距离是基于物种系统进化关系进行计算的样本间的距离, 只考虑了物种的有无。Weighted Unifrac距离是 结合OTU的丰度信息和系统进化关系获得的样本间的距离。 Unweighted Unifrac距离对稀有物种比较敏感, 而Bray Curtis和Weighted Unifrac距离则对丰度较高的物种更加敏感。 Weighted Unifrac距离,Unweighted Unifrac距离及Bray Curtis距离 作为Beta多样性距离是衡量两个样品间的相异系数的指标, 其值越小,表示这两个样品在物种多样性方面存在的差异越小。 图3-4-1为不同样品之间以上三种距离热图的展示。 详细的数值可以查看结果目录 4-BetaDiversity/1-BetaDiversitySummary/ 。

图3-4-1 Beta多样性指数热图
说明:热图方格的颜色代表样品两两之间的相异系数, 相异系数越小的两个样品,物种多样性的差异越小。 从上到下,在热图中分别代表Bray Curtis距离, Weighted Unifrac距离和Unweighted Unifrac距离。
Beta多样性NMDS及PCoA的分析,我们提供了qiime2 [1] 及R语言Phyloseq包 [8] 两种可视化结果。主坐标分析(PCoA),是一种与PCA类似的降维排序方法, 从多维数据中提取出最主要元素和结构能够提取出最大程度 反映样品间差异的三个坐标轴,从而将多维数据的差异反映在三维坐标图上, 进而揭示复杂数据背景下的简单规律。 区别在于PCA是基于样品的相似系数矩阵来寻找主坐标, 而PCoA是基于距离矩阵来寻找主坐标。我们基于Bray Curtis距离、 Weighted Unifrac距离和Unweighted Unifrac距离来进行PCoA分析, 并选取贡献率最大的主坐标组合进行作图展示,图中样品的距离越接近, 表示样品的物种组成结构越相似。分析结果文件夹分别提供了PCoA的2D和3D图, 方便选择使用。
图3-4-2 基于Unweighted Unifrac的PCoA 3D图 (图片可拖动旋转,可调色)
说明:横坐标(Axis 1)表示第一主成分, 百分比则表示第一主成分对样品差异的贡献值;纵坐标(Axis 2) 表示第二主成分,百分比表示第二主成分对样品差异的贡献值; Axis 3坐标表示第三主成分,百分比表示第三主成分对样品差异的贡献值。 客户可以根据元数据的分组信息可以个性化展示其他分类的距离关系。 举例:可在右边设置区域的Select a Color Category 下拉单选择分组信息,则同一个组的样品使用同一种颜色表示。
其他距离完整的PCoA展示见结果目录 4-BetaDiversity/2-PCoA/
无度量多维标定法(NMDS)统计是一种适用于生态学研究的排序方法, 类似于PCoA,通过样本的分布,或者基因的分布,可以看出组间差异, 组内差异等。NMDS包括一类排序方法,其设计目的是为了克服以前的排序方法 (即线性模型,包括PCoA)的缺点,NMDS的模型是非线性的, 能更好地反映生态学数据的非线性结构。 非度量多维尺度法是一种将多维空间的研究对象(样本或变量)简化到 低维空间进行定位、分析和归类, 同时又保留对象间原始关系的数据分析方法。 适用于无法获得研究对象间精确的相似性或相异性数据, 仅能得到他们之间等级关系数据的情形, 图3-4-3为基于Unweighted Unifrac距离进行的NMDS分析。

图3-4-3 2D NMDS分析
说明:图中的点代表样本,不同颜色的点属于不同样本(组), 点与点之间的距离表示样本菌群差异程度,两点之间的距离越近, 表明两个样本之间的微生物群落结构相似度越高,差异越小。 其他距离NMDS展示见结果目录 4-BetaDiversity/3-NMDS/ 。
PLS-DA的分析由R语言mixOmics包 [9] 完成。PLS-DA分析, 即偏最小二乘法判别分析,是一种用于判别分析的多变量统计分析方法, 根据观察或测量到的若干变量值,来判断研究对象如何分类。 PLS-DA属于有监督的分析方法,需要将检测样本按照类别进行分组, 在计算数学模型时把各组加以区分,忽略组内的随机差异,突出组间系统差异, 在观测因子远大于样本数量时具有较好的样本区分表现, 但这往往是过拟合现象造成的,即PLS-DA模型在当前数据集表现很好, 但如果把这个模型用于其它样本的预测,可能区分效果就不好了, 增加样本量有助于减轻过拟合。 同时PLS-DA会选择性忽略在分组间没有差异的微生物, 所以其坐标图比PCoA图分的开,但是并不能代表微生物群落整体的情况, 只保留了分组间有差异的信息。基于所有丰度大于10的OTU, 我们做了PLS-DA分析,提供了坐标图,ROC曲线, VIP(variable importance in projection)表格,请参见结果目录 4-BetaDiversity/5-PLS-DA/ 。
这里对PLS-DA分析结果说明如下:
- PLS-DA坐标图每个点代表一个样本, 颜色相同的点属于同一分组,相同分组的点以椭圆标出。 如果属于同一分组的样本彼此之间距离越近, 同时不同分组的点之间的距离越远,表明分类模型效果越好。
- 在ROC曲线图中,右侧列出了各分组和其他所有分组进行判别分析的情况下, 所得到的曲线下面积(AUC),该值越接近于1,说明区分效果越好, 有多个微生物在分组间丰度有差异。
- 同时,VIP表格中的VIP (comp.1和comp.2相关性很大,看comp.1或comp.2都行, comp.1是区分效果最好的成分,comp.2次之,可重点看comp.1), 体现了各个微生物在判别分析中的重要性,该值越大, 表明该微生物在分组间差异越大。 VIP大于1的微生物应该是重点关注的微生物。
3.4.2 基于Beta多样性指数的组间菌群结构比较
在得到整体的Beta多样性指数之后, 接下来结合分组信息来用PERMANOVA方法和ANOSIM方法比较 在各个样品分组之间的微生物组成结构是否有显著性差异。 PERMANOVA和ANOSIM的分析基于qiime2 "qiime diversity beta-group-significance"命令。
基于Unweighted Unifrac距离进行的PERMANOVA分析
说明:上面的箱线图横向为分组信息, 各个分组名字后面括号内的n值代表在组间的比较次数。 纵向为比较的样品之间的距离。有多少个分组就会有多少个箱形图, 以上图仅展示了所有分组对其中第一个分组的样品之间的距离绘制的箱形图, 其他组的详细信息请查看结果文件。 下面的表格是箱线图详细数据说明表格,q值是校正过的p值, 更具参考价值,q<0.05可推断组间菌群结构存在显著差异。
基于其他距离的菌群结构差异显著性分析结果可以查看结果目录 4-BetaDiversity/6-GroupSignificance/ 。
3.5 系统进化分析
进化树的绘制由R语言ggtree包 [10] 完成。选取关注的OTU代表序列进行系统进化分析 (每个属选取一个丰度最高的OTU作为代表OTU, 之后再选取丰度最高的前50个属)绘制进化关系树图, 同时结合OTU的在各个分组的绝对丰度进行热图可视化展示(图3-5-1)。 客户可以点击 5-Phylogenetics/ 查看完整信息。

图3-5-1 系统发育进化树及组间丰度分布热图
说明:左边为进化树,不同颜色的分支代表不同的门, 每条末端的树枝代表一个OTU,末端注释了对应OTU所属的属分类, 如果没有对应的属分类,则以unclassied_genus表示, 右边热图是标准化后的丰度,值越大代表相对丰度越高。 丰度标准化:每个样本的绝对丰度减去该物种绝对丰度的平均值 再除以标准差,从而使得标准化之后的丰度平均值为0,标准差为1。
3.6 关联统计分析
6-AssociationAnalysis/
├── 1-RDA
│ └── [物种分类水平]
│ ├── [分组方案名]_RDA.envfit.xls [RDA各环境因子坐标,解释方差,p值]
│ ├── [分组方案名]_RDA.Factors.PERMANOVA.xls [所有环境因子对微生物群落变异的解释方差的p值计算结果]
│ ├── [分组方案名]_RDA_features_location_plot.svg [RDA微生物点图]
│ ├── [分组方案名]_RDA.features.xls [RDA微生物点坐标]
│ ├── [分组方案名]_RDA.Group.PERMANOVA.xls [分组方式对微生物群落变异的解释方差的p值计算结果]
│ ├── [分组方案名]_RDA_sample_location_plot.svg [RDA样本点图]
│ ├── [分组方案名]_RDA_sample_location_plot_with_labels.svg [RDA样本点图(带样本名)]
│ ├── [分组方案名]_RDA.sample.xls [RDA样本点坐标]
│ └── DCA_output.xls [DCA分析各排序轴值]
├── 2-CorrelationHeatmap
│ └── [物种分类水平]
│ ├── Correlation_heatmap.svg [相关性热图]
│ ├── p_value_matrix.xls [相关系数p值矩阵]
│ └── spearman_rank_correlation_matrix.xls [spearman相关系数矩阵]
└── 3-NetworkAnalysis
└── [物种分类水平]
├── edges.xls [network.html中连线数据, 可作为CytoScape输入]
├── network.html [交互式网络图, 请结合右侧参数, 调整和理解]
├── nodes.xls [network.html中节点数据, 可作为CytoScape输入]
├── Correlation_network.svg [相关性网络图]
├── p_value_matrix.xls [相关系数p值矩阵]
└── spearman_correlation_matrix.xls [spearman相关系数矩阵]
3.6.1 CCA/RDA分析
CCA/RDA的分析主要依赖R语言vegan包[11], 以及用ggplot2[12]进行可视化。 CCA/RDA(DCA判断用哪一种分析)分析是基于对应分析发展的一种排序方法, 将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析。 RDA是基于线性模型,CCA是基于单峰模型。 本报告先进行DCA分析,看最大轴的值是否大于4,如果大于4.0, 就选CCA,否则选RDA。该分析主要用来反映微生物群落与环境因子之间的关系, 可以检测环境因子、样品、微生物群落(或功能)三者之间的关系或者两两之间的关系, 可得到影响样品分布的重要环境驱动因子。该分析给出的所有p值都是反映解释变量 (连续的环境因子,或者分组方案)对微生物群落变异的解释程度是否显著 (简单的说就是解释变量对微生物群落是否有影响,影响是否显著), 所有p值都是用R语言vegan包里的置换检验得出的 (permutation test),*_features_location_plot图中的p值 反映了所有连续的数值变量(环境因子)对微生物差异的解释程度(总的p值), 表格*_RDA.envfit中的p值反映了每个环境因子对微生物差异的解释程度, *RDA_sample_location_plot图中的p值反映了分组对微生物 差异的解释程度,p<0.05, 解释方差显著。

图3-6-1 属水平微生物CCA/RDA排序图
在CCA/RDA物种排序图内,环境因子用箭头表示, 箭头连线的长度代表某个环境因子与群落分布和种类分布间相关程度的大小(解释方差的大小), 箭头越长,说明相关性越大,反之越小。箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小, 夹角越小,相关性越高;反之越低。环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系, 钝角时呈负相关关系。每个点代表一个物种,点越大,对应物种丰度越高, 灰色点代表丰度较低的物种,未在图中注释物种名称,将物种投影到各个环境因子, 对应的值即为该物种倾向于存在的环境(喜欢的环境)。或者说,将物种点与原点连线, 物种间,物种与环境因子间,环境因子间的夹角的余弦值近似于相关系数,至于有多近似, 就要看RDA1/CCA1和RDA2/CCA2两个坐标轴的解释方差百分比有多大,越大越近似。 对于样本排序图,样本点之间的距离近似于微生物群落结构差异程度, 样本点投影到环境因子对应的值近似于该样本真实的环境因子值。

图3-6-2 CCA/RDA样本排序图
环境因子用箭头表示, 箭头连线的长度代表某个环境因子与群落分布和种类分布间相关程度的大小(解释方差的大小), 箭头越长,说明相关性越大,反之越小。箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小, 夹角越小,相关性越高;反之越低。环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系, 钝角时呈负相关关系。样本点之间的距离近似于微生物群落结构差异程度, 样本点投影到环境因子对应的值近似于该样本真实的环境因子值。
*_RDA.envfit.xls表格各列解释如下:
- 环境因子
- RDA1: 表征环境因子的方向,与横轴正方向夹角的余弦值
- RDA2: 表征环境因子的方向,与横轴正方向夹角的正弦值
- r2: 环境因子对微生物群落变异的解释方差
- p value: 置换检验计算的解释方差的p值,小于0.05,可推断该环境因子对微生物群落影响显著
*_RDA.*.PERMANOVA.xls表格为所有环境因子(或分组方式)对微生物群落结构变异的解释方差的p值计算结果, 用的是PERMANOVA置换检验的方法,各列解释如下:
- 方差组分(模型部分,剩余部分)
- Df: 自由度
- Variance: 方差剖分
- F: PERMANOVA检验统计量
- Pr(>F): PERMANOVA检验p值
3.6.2 相关性热图分析
如果提供了环境因子的数据,比如 pH值、温度值、临床因子等,可以进行相关性热图分析
相关性热图的绘制主要运用了R语言pheatmap包 [13]。 相关性热图可以用于分析环境因子或其他临床表型数据与微生物群落或物种之间是否显著相关, 然后计算环境因子与微生物物种间的Spearman相关系数,并用热图展示。 该分析可以挑选出与某种环境因子或疾病显著相关的物种。

图3-6-3 属水平微生物与表型之间的相关性热图
X轴上为环境因子,Y轴为物种。通过计算获得R值(秩相关)和p值。 R值在图中以不同颜色展示,p值若小于0.05则用 * 号标出, 右侧图例是不同R值的颜色区间,同时,左边的色条标注了物种所属门分类。 * 0.01≤ p <0.05,** 0.001≤p < 0.01,*** p < 0.001。 先依据显著的环境因子个数排名(越多越靠前),再依据显著的p值平均值排名(越低越靠前), 该图只展示排名前20个微生物,如果没有显著相关的微生物,则无法作图。
其他分类水平的相关性热图请查看 6-AssociationAnalysis/2-CorrelationHeatmap
3.6.3 物种相互作用网络分析
相关网络主要运用R语言igraph包 [14] 绘制。基于物种样本间的相对丰度进行Spearman获得样本内 或样本组内的物种之间的相互关系, 然后用展示物种间相互关系的可视化软件进行物种相互作用网络构建(图3-6-4)。 该分析可以寻找相互拮抗或协同的物种, 从而寻找环境样本中的微生物群体协同或相互抑制信息, 例如针对某种疾病相关的微生物群落,就可以发现共同作用的微生物或相互抑制的微生物, 为其治疗做指导。同时我们也提供基于相对组成成分数据的相关性计算, 制作了可动态调整的相关性网络 网页报告。

图3-6-4 属分类水平物种相互作用网络图
说明:样本组各自富集的物种的相互作用network图的比较, 圆圈代表一个物种,大小代表其相对丰度, 不同的颜色代表不同的物种门分类, 圆圈之间的线条代表这两个物种间的相关性显著(p值小于0.05), 线条颜色红色代表正相关,蓝色代表负相关,线条越粗,相关系数绝对值越大。
其它分类水平的相关网络图请查看 6-AssociationAnalysis/3-NetworkAnalysis
我们也提供了交互式的网络图(network.html), 交互式网络图,可以通过右侧的参数调整连线粗细, 连线颜色,节点颜色,节点大小等对应的意义,可调整网络布局, 获取网络拓扑参数
nodes.xls各列如下:
- node: feature名, 如物种名等
- feature_type: 输入表最后一列feature分类, 如门水平分类, 可调整network.html对应到节点颜色
- sum_abundance: feature所有样本总丰度, 可调整network.html对应到节点大小
edges.xls各列解释如下:
- source: feature1名称, 如物种名等
- target: feature2名称, 如物种名等
- correlation: feature1和feature2的相关系数,
- absolute_correlation: feature1和feature2的相关系数绝对值, 可调整network.html对应到连线粗细
- p-value: feature1和feature2的相关系数p值
- -log10p: -log10(feature1和feature2的相关系数p值), 可调整network.html对应到连线粗细
- correlation_type: negative表示负相关, positive表示正相关, 可调整network.html对应到连线颜色
3.7 功能预测分析
7-FunctionAnalysis/ ├── 1-Barplots │ ├── bar-plots-top20 │ │ ├── [功能数据库名称]_barplot.svg [总丰度前20功能百分比堆积柱形图] │ │ └── [功能数据库名称]_table.xls [总丰度前20功能百分比堆积柱形图对应数据表] │ └── bar-plots-top20-group-mean │ ├── [功能数据库名称]_barplot.svg [总丰度前20功能分组平均丰度百分比堆积柱形图] │ └── [功能数据库名称]_table.xls [总丰度前20功能分组平均丰度百分比堆积柱形图对应数据表] ├── 2-PCAPlots │ ├── [功能数据库名称]_PCA_3D.svg [功能3D PCA图] │ ├── [功能数据库名称]_pca_points_ordinates.xls [PCA 坐标表] │ └── [功能数据库名称]_PCA.svg [功能2D PCA图] ├── 3-SignifcanceAnalysis │ ├── 1-DunnTest │ │ └── [功能数据库名称]_[分组方案名称]_DunnTest.xls [DunnTest多重比较结果表] │ └── 2-ANOVA_And_Duncan │ ├── [功能数据库名称]_[分组方案名称]_all_pathway_anova_results.xls [anova比较结果表] │ ├── [功能数据库名称]_[分组方案名称]_all_significant_pathway_barplot_of_duncan.svg [duncan多重比较结果图] │ ├── [功能数据库名称]_[分组方案名称]_all_significant_pathway_duncan_results.xls [duncan多重比较结果表] │ └── [功能数据库名称]_final_data_for_anova.xls [anova输入数据] ├── [功能数据库名称].pathway.abundance.xls [相应数据库功能丰度表] ├── OTU.[基因家族].abundance.xls [OTU各基因家族的基因数目(不是丰度)] └── Sample.[基因家族].abundance.xls [基因家族丰度表]
3.7.1 预测微生物群落功能
PICRUSt2 [15] 的原理是基于已测微生物基因组的序列, 推断它们的共同祖先的基因功能谱, 对数据库中其它未测物种(基因组未知)的基因功能谱进行推断, 构建微生物全谱系的基因功能预测谱, 最后,将测序得到的微生物组成映射到数据库中, 对微生物代谢功能进行预测。 图3-7-1展示的是预测得到的MetaCyc数据库通路的百分比组成柱形图。 对于16S扩增子,我们还提供了KEGG数据库的通路预测结果。 KEGG功能根据注释深度可以分为三个分类层级, 类似于微生物的分类层级。什么是分类层级? 比如我们知道奶牛的数量,知道肉牛的数量, 我们把奶牛的数量和肉牛的数量加起来得到了牛的数量, 这里的奶牛和肉牛相当于L3分类层级,牛相当于L2分类层级, 如果再把猪和牛的数量加起来得到脊椎动物的数量,脊椎动物相当于L1层级, 以此类推。从L1到L3相当于一个细化分类的过程。看哪一个级别的结果比较好? 通常看L3级别的结果,兼顾L3级别所属的L1级别分类。 详细结果请查看 7-FunctionAnalysis/1-Barplots/ 。

图3-7-1 MetaCyc通路预测组成柱形图
说明:纵轴表示注释到某类型的功能的相对比例;横轴表示分组名称; 各颜色区块对应的功能类别见右侧图例。
3.7.2 预测功能PCA分析
图3-7-2是基于MetaCyc数据库的功能(通路)PCA图。 对于16S扩增子,我们也提供了KEGG数据库通路的PCA。 详细结果请查看 7-FunctionAnalysis/2-PCAPlots/ 。

图3-7-2 MetaCyc通路的PCA分析结果展示
说明:横坐标(x)表示第一主成分, 百分比则表示第一主成分对样品差异的贡献值; 纵坐标(y)表示第二主成分,百分比表示第二主成分 对样品差异的贡献值;图中的每个点表示一个样品, 同一个组的样品使用同一种颜色表示;如果不同分组的样本分布 在不同区域,则说明不同分组的预测功能组成差异较大。
3.7.3 组间功能差异性分析
在得到功能注释后,结合分组信息, 利用ANOVA+Duncan和Dunn test分析了微生物群落预测功能在组间 是否有显著性差异。Dunn test分析基于R语言dunn.test包 [16] ,其中,结果文件中KW_pvalue, DunnTest_comparison, DunnTest_Z, DunnTest_PValueAdjusted 分别为Kruskal-Wallis检验P值, DunnTest多重比较的分组组合,DunnTest的检验统计量, DunnTest的校正后P值。ANOVA+Duncan基于R语言agricolae包 [17] ,在进行ANOVA+Duncan检验之前, 我们先筛除观测样本小于总样本50%的代谢通路 (Filter the pathways observed in less than 50% of total samples), 再基于相对丰度进行差异分析,图3-7-3是用ANOVA和Duncan检验得到的 所有显著差异的MetaCyc通路,当且仅当ANOVA分析校正“错误发现率”后的p值 (p.adj)小于0.05时,才会进一步做Duncan检验,并绘图。 对于16S扩增子,结果文件夹中也提供了根据KEGG L1水平,分别画的图, 以减小一个图的面积。 点击此处查看功能差异性分析详细结果 。

图3-7-3 ANOVA和Duncan检验得到的所有显著差异的MetaCyc通路
说明:横坐标为通路名字;对于每个通路, 用不同颜色表示不同的分组,两个分组上方如果有相同的字母, 则代表差异不显著,否则差异显著。
写在最后:如果您对图片中的字形,字号,字的内容 (包括样本名称,分组名称),字的方向,图片中某个元素的位置,大小,方向, 线条粗细等不涉及统计方法的方面不满意,或者想删除图中的某些元素, 推荐使用AI软件(Adobe Illustrater)进行修改,所有的矢量图, 包括pdf和svg,都是可以用AI进行排版和编辑的。 祝发文顺利!
四 分析所用软件的版本
| 软件 | 版本 |
|---|---|
| QIIME | 2022.2 |
| DADA2 | 1.22.0 |
| Greengenes2 | release 2022.10 |
| SILVA | release 138 |
| LEfSe | 1.0.8 |
| DESeq2 | 1.26.0 |
| phyloseq | 1.30.0 |
| mixOmics | 6.10.9 |
| ggtree | 3.3.1 |
| vegan | 2.6.0 |
| ggplot2 | 3.3.5.9000 |
| pheatmap | 1.0.12 |
| igraph | 1.2.4.2 |
| PICRUSt2 | 2.3.0 |
| dunn.test | 1.3.5 |
| agricolae | 1.3.3 |